(Updated on 2016/1/28)
理化学研究所分子スケール研究開発チーム
チームリーダー 木寺詔紀
タンパク質や核酸、薬剤など生体分子を計算対象とする。反応の時間スケールが遅い(ミリ秒~秒)ために、通常のMDでは追跡が困難な、生体分子同士の相互作用やこれに伴う立体構造変化過程の再現を目的とする。
マルチコピー・マルチスケール連成分子動力学法
レプリカ交換法、MSES法、ストリング法、粒子フィルタ
コピー間並列(MPI並列)、ループ分割(MPI・スレッド並列)
C++, MPI, OpenMP, LAPACK, FFTW, NetCDF
ソース・コードをISLiMダウンロードサイトから公開済み
- アデニル酸キナーゼ立体構造変化パスウェイ探索
- 1,600万原子系(6万原子系×256コピー)
- RICC 8192コア、メモリ容量 52 GB、ディスク容量 10 TB
- 多剤排出トランスポーター(AcrB)
- 1億2,800万原子系 (50万原子系×256コピー)
- 65,536コア(256コア/コピー)使用
- メモリ容量 416 GB、ディスク容量 1 PB
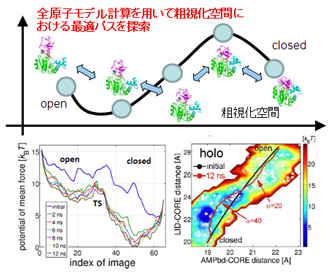
図. ストリング法の概念図(上)とタンパクへの応用例(下)
- 薬剤などの生理活性物質がタンパク質など生体高分子に結合する過程を再現できるようになる
- 薬剤の結合やタンパク質同士の結合に伴う立体構造変化を予測できるようになる
- 結合過程や立体構造変化過程の解析から、活性の高い物質の創出やタンパク質の機能改変につながる手掛かりが得られる。
理化学研究所基幹研准主任研究員 杉田有治
タンパク質のまわりの環境を含めたタンパク質の構造予測や、構造変化予測、自由エネルギー計算等を効率良く行なう為のプログラム。多次元レプリカ交換分子動力学計算を行なう。「京」に対応したMarbleを分子動力学計算に用いるが、その他のプログラムにも対応可。
古典粒子(MD)
レプリカ交換法
複数のレプリカの並列実行
Fortran90, C, MPI
ソース・コードをISLiMダウンロードサイトから公開済み
(AAQAA)3タンパク質(1万原子系)をRICCの8192コアで解析
- タンパク質の構造変化、構造予測および、タンパク質間の結合自由エネルギープロファイル
- 1-100万原子系を64万コアで解析
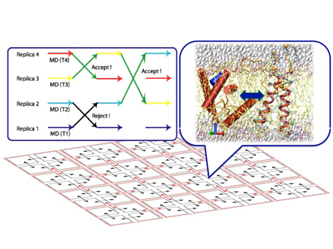
レプリカ交換MD法によるタンパク質の分析
- 膜蛋白質や大規模な系の構造変化予測や、これまでより大きな系での構造予測ができる。
- 原子解像度でタンパク質の複合体形成機構の理解が深まる。
- 例えば応用として、タンパク質と疾患との関係等の理解が深まり、将来の医学や創薬に役立つ事が期待できる。
横浜市立大学大学院総合理学研究科准教授 池口満徳
タンパク質や核酸など生体分子の立体構造で、水素まで含んだ全原子モデルを解く。
古典分子動力学
- FFTを用いたParticle Mesh Ewald法
- シンプレクティック数値積分法
空間分割,スレッド並列はループ分割
C, MPI, OpenMP
ソース・コードをISLiMダウンロードサイトから公開済み
- 10万-100万原子系
- Cray XT6で3000コア並列
- メモリ容量 4 GB/node、ディスク容量 1 TB
- 多剤排出トランスポーター(約50万原子)
- 反復回数109回以上
- メモリ容量 16 GB/node、ディスク容量 10 TB
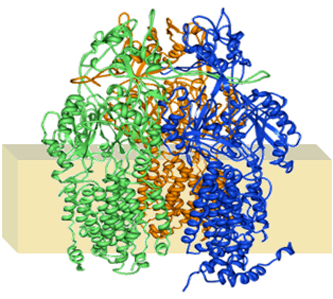
多剤排出トランスポーターAcrB
- 膜タンパク質が大規模な構造変化を行う様子をシミュレートすることができる。
- 薬剤耐性菌の原因因子である多剤排出トランスポーターのシミュレーションを行うことにより、菌が異物を排出する際の分子機構を明らかにする。
- 「MARBLE-K」利用講習会 -> リンク
京都大学理学研究科准教授 高田彰二
粗視化分子モデル計算により大規模生体分子の長時間シミュレーションを行う。
粗視化された分子モデルによる古典分子動力学法
Langevin方程式の時間発展を数値的に積分する。
Neighbor list方式、レプリカ交換法
Fortran90, MPI, OpenMP
ソース・コードをISLiMダウンロードサイトから公開済み。ただし生体膜、核酸については要相談。
- 1万残基タンパク質のミリ秒相当のシミュレーションが可能
- PCクラスタで8192コア並列
- メモリ容量 2 GB/コア、ディスク容量 1 TB
- 10万粒子(100万原子)系の秒相当のシミュレーション
- 反復回数1010回以上
- メモリ容量 2 GB/コア、ディスク容量 1 PB
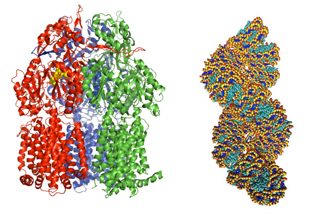
トランスポーター(左)とDNAヒストン複合系(右)
- X線回折やNMRによる構造情報が存在するタンパク質、核酸などの動態の分子動力学シミュレーション。とくに構造変化を伴うタンパク質ドッキング、分子モーターやトランスポーターの大規模構造変化などをシミュレーションできる。
- 例として、キネシンの動態、多剤排出トランスポーターの動態、DNAヒストン複合系(ヌクレオソーム)など。
東京大学生産技術研究所教授 佐藤文俊
- 複雑かつ大規模・高精度が要求されるタンパク質の全電子を密度汎関数法に基づきカノニカル軌道で計算する。
- 励起状態も含めた第一原理分子動力学計算が可能。
分子軌道法
直接法による密行列の対角化
原子分割法、RT アルゴリズム+シェルタイプ分類均等分割法。
C++, MPI, OpenMP, ScaLAPACK
- ISLiMダウンロードサイトから公開済み
- 306 残基、27,000 軌道、基底状態計算(世界最大)
- Altix3700(64 CPU)で実行、300 GFlops
- メモリ容量 256 GB、ディスク容量 1 TB
- ほぼ全てのタンパク質に対応するため計算規模を3倍に
- 基底状態だけでなく、励起状態のダイナイミクスを解析
- メモリ容量 2PB 、ディスク容量 10 PB(励起状態)

インスリン6量体静電ポテンシャルの古典計算との差
- 創薬の信頼性の高い基礎研究のみならず、医薬品開発の高品位化および高効率化、ならびに次世代の医薬品研究開発モデルの創成といった応用に貢献できる。
- 方法特許やビジネスモデル特許などを含め欧米に対する優位性を実現できる。
- 触媒、分子素子、環境物質などへの応用も大いに期待できる。
- 「ProteinDF」利用講習会 -> リンク
京都大学大学院理学研究科准教授 林重彦
量子化学的手法(QM)と分子力場法(MM)のハイブリッド であるQM/MM 法により、生体分子内の反応基質分子の自由エネルギー最適構造の決定及び反応性解析を行う。
分子軌道法、分子力場法
Reweighting法(自由エネルギー)、 Ewald 法(クーロン)。
MM 構造サンプル分割
FORTRAN77, GAMESS の socks ライブラリ
ソースコードを無償で利用可能。要相談。
- a-amylase 酵素における polysaccharide(二量体部分)の自由エネルギー構造最適化。
- 7万原子系、QM 原子数 69、基底関数 650、MM 構造サンプル数140,000
- PC128 コア、メモリ容量 256 GB、ディスク容量 50 GB
- 多剤排出トランスポーター
- 約50万原子、MM構造サンプル数100 万
- メモリ容量 4 TB、ディスク容量 4 TB
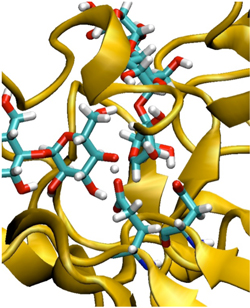
a-amylase 酵素中のpolysaccharide 基質の加水分解反応遷移状態
- 酵素反応における基質の反応性解析が行える。
- 生体エネルギー変換で最も重要であるプロトン濃度勾配を用いた分子機能が明らかになる。
大阪大学蛋白質研究所教授 中村春木
分子軌道法および密度汎関数理論(DFT)を用いた蛋白質等分子系の全電子第一原理計算を行う。
分子軌道法、密度汎関数理論、Hartree-FockとのHybrid DFT
多中心Euler-Maclaurin-Lebedevの求積分法、固有値問題等
MPI並列化(ハイブリッド並列化へ拡張中)
Fortran77, Fortran90, MPI, HDF, ScaLAPACK, LAPACK等
ソース・コードをISLiMダウンロードサイトから公開済み
- バクテリオクロロフィル等の色素
- 数百~千電子系をPCクラスタ8192コアで解析
- メモリ容量 1.2GB/コア、ディスク容量 100 MB
- 数100~数万電子系の基底状態/励起状態の電子構造を64万コアで解析
- メモリ容量 1.6GB/コア、ディスク容量数GB
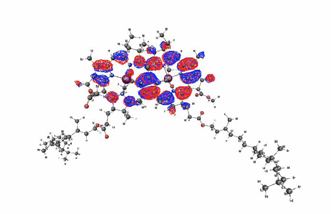
Rhodobacter sphaeroides由来の光合成活性中心の色素(バクテリオクロロフィルaダイマー)のHOMO軌道
- 遷移金属を含む蛋白質系の基底状態および励起状態の電子構造の解明
- 電子構造解析による反応機構の解明
- 磁気的相互作用など、蛋白質構造が不明な場合のプローブ(EPR等)と付けられるパラメータの計算
大阪大学蛋白質研究所教授 中村春木
電子状態の効果を分子動力学に取り込むことで酵素反応機構などの生体高分子の精密解析を行う。
分子軌道法、分子動力学法
直接法による密行列対角化、反復法による疎行列連立方程式求解、改良Wolf等
領域分割(分子軌道法)、原子分割(MD)
FORTRAN77, Fortran90, C, MPI, OpenMP, ScaLAPACK, LAPACK
未公開
- プロリン及びN-metylacetamide(量子系)の水溶液中(水分子は古典粒子)における異性化熱力学過程の解明
- 20000原子を蛋白研PCクラスタ256コアで解析
- メモリ容量 300Mbyte/コア、ディスク容量 3GB
- 数100-数1000電子系と、それを取り巻く数10万古典原子による反応自由エネルギー地形を64万コアで解析
- 基底状態だけでなく、励起状態のダイナイミクスを解析
- メモリ容量 1.6GB/コア 、ディスク容量数GPB
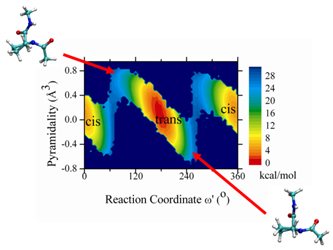
ピラミダリティの反応座標依存性の量子古典連成計算
- 生命活動に重要な酵素反応機構や、その反応を制御する薬物などの開発に有用な知見を得ることができる。