(Updated on 2013/3/4)
Akinori KIDERA, Team Leader, Molecular Scale R&D Team, Computational Science Research Program (CSRP), RIKEN
Platypus-MM/CG targets biomolecules, such as proteins, DNAs, and drugs. It aims to simulate the interactions between biomolecules and the involved 3D conformational changes, whose time scale is too long (msec to sec) to be solved with the conventional MD methods.
Multi-copy, multi-scale coupled molecular dynamics (MD)
Replica-exchange method, Multi-scale essential sampling method (MSES), On-the-fly string method, and Particle filters
MPI among copies, and MPI-thread hybrid parallelization for the divided loops
C++, MPI, OpenMP, LAPACK, FFTW, and NetCDF
Source code is available through ISLIM download site.
- 3D conformational changes and pathway searches for adenylate kinase
- 16-million-atom system (60,000-atom system x 256 copies)
- 8,192-core parallel computing with RIKEN RICC system
- Required memory/disk storage: 52 GB/10 TB
- Multi-drug efflux transporter AcrB
- 1-billion-atom system (500,000-atom system x 256 copies)
- Parallel computing with 65,536 cores (256 cores/copy)
- Required memory/disk storage: 3.3 TB/1 PB.
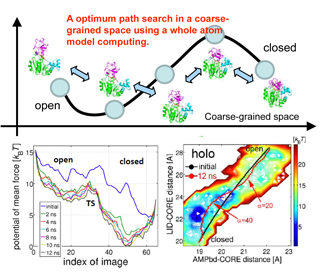
Figure 1. The concept of on-the-fly string method (upper), and the application results for a protein (lower).
- Simulation of processes for biologically active agents like drug to dock biomacromolecules like protein
- Prediction of 3D conformational changes in docking for drugs and in coupling of proteins
- Clues leading to discovery of bioactive materials or function modified proteins based on above simulations.
Yuji SUGITA, Associate Chief Scientist, RIKEN Advanced Science Institute
The code computes multidimensional replica exchange MD to efficiently predict protein conformations with surrounding environment, conformation change, free energy values. The code uses MARBLE, which will run on the K computer, for MD calculation. Other MD codes can be applicable to Platypus-REIN
Classical particles (MD)
Replica exchange method
Parallel execution of multireplica
Fortran90, C, MPI
Source code is available through ISLIM download site.
(AAQAA)3 protein (10-thousand-atom system) run on RICC's 8,192 cores
- Protein's conformation change and conformation prediction.
- Binding free energy profile between proteins
- Analysis of 1-million-atom system with 640 thousands of cores.
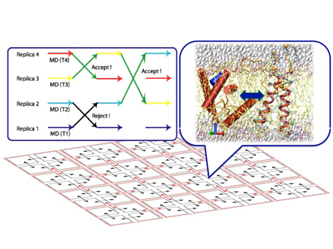
Figure 1. Analysis of a protein by the replica exchange method.
- Predicting the conformation or conformation change for membrane proteins and unprecedented largest proteins
- Deeper understanding about the mechanism for forming protein complexity in the atomic scale resolution
- As an application, the deep understanding about protein-disease relationship can help medicine and drug discovery in the future.
Mitsunori IKEGUCHI, Associate Professor, Supramolecular Biology, Yokohama City University
The code performs all-atom biomolecular simulations for protein, DNA, and membranes.
Classical all-atom molecular dynamics simulations
- Particle Mesh Ewald method
- Symplectic numerical integration method
Domain decomposition. Thread parallelization
C, MPI, OpenMP
Source code is available through ISLIM download site.
- A 100-thousand to 1-million-atomic system
- Cray XE6's 3,000 cores
- Required memory/disk storage: 4 GB per node/1 TB
- Multidrug efflux transporter (about 500 thousand atoms)
- Iterations: >109
- Required memory/disk storage: 16 GB per node/10 TB.
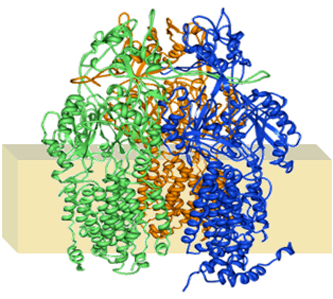
Figure 1. Multidrug efflux transporter AcrB
- Simulations for large biomolecules with explicit membrane and solvent molecules
- By using simulations of the multidrug efflux transporter that causes drug resistance, the molecular mechanism underlying multidrug efflux will be elucidated.
Shoji TAKADA, Associate Professor, Kyoto University
Using the coarse-grained molecular model, the code does a long simulation of large biomolecules
The coarse-grained classical MD model
Time integration of the Langevin equation (differential equation)
Neighbor list approach, replica exchange
Fortran90, MPI, OpenMP
Source code is available through ISLIM download site.
- A several-millisecond simulation for the 10 thousands of residues of the protein
- Use 8,192 cores of the x86 cluster
- Required memory/disk storage: 2 GB per core/1 TB
- A simulation for the process of milliseconds to seconds of the 100 thousands of particles (1 million atoms)
- Number of iterations: >1010
- Required memory/disk storage: 2 GB per core/1 PB.
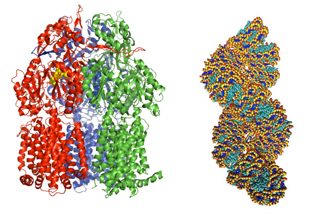
Figure 1. A transporter (left) and a DNA-histone complex (right).
- The MD simulation for the dynamic behaviors of the proteins and DNAs for which structural data are available via the X-ray diffraction or NMR. In particular, the dynamic behaviors of protein complex, molecular motors, and transporters that exhibit large conformation changes can be simulated
- The typical examples are the dynamic behaviors for the kinesine motor, the multidrug efflux transporter, and the DNA-histone complex.
Fumitoshi SATO, Professor, the University of Tokyo
- The code solves the all-electron canonical wave functions of a protein, which requires complicated, large scale, and highly accurate computation, based on the density-functional approach.
- The code can do the first principle MD computation including excitation states.
The molecular orbital (MO) method
The Roothaan-Hall minimization (diagonalization) in the canonical MO basis by the direct method
RT algorithm + shell-type-classification equal-partitioning method
C++, MPI, OpenMP, ScaLAPACK
- Source code is available through ISLIM download site.
- The world-largest 306 residues 27,000 orbitals ground-state computation (Fig. 1)
- With Altix3700(64 CPU, 300 GFlops)
- Required memory/disk storage: 256 GB/1 TB
- 20,000 atoms calculation in the ground states, which covers the most proteins
- Dynamic analysis
- Excitation states
- Required memory/disk storage: 2 PB/10 PB (Excitation state).

Figure 1. The differece electrostatic potential in the insulin hexamer between the ProteinDF and a classical computation.
- The code contributes to not only the highly reliable basic research in the drug discovery but also more highly qualified and efficient drug development as well as the creation of next-generation R&D models in the pharmaceutical.
- It can bring the Japan's superiority in patents over foreign countries.
- It can apply to catalysts, molecular elements, and environmental materials.
Shigehiko HAYASHI, Associate Professor, Kyoto University
The code determines the free-energy optimal conformation in the reactive subtrate molecule in the biomolecules, and analyzes its reactivity by the hybrid approach with the quantum chemistry method (QM) and the molecular field method (MM).
The molecular orbital method, the molecular force field method
Reweighting method (Free energy), Ewald method (Coulomb)
Conformation sample decomposition for MM
FORTRAN77, the socks library in GAMESS
To be released.
- The free-energy optimal conformation for polysaccharide (dimer portion) of the a-amylase kinase
- A 70-thousands-of-atoms system, QM atoms: 69, base functions: 650, MM conformation samples: 140,000
- Required memory/disk storage: 256GB/50GB (128 cores)
- Multidrug efflux transporters
- About 500 thousands of atoms, 1-million MM-conformation samples
- Required memory/disk storage: 4 TB/ 4 TB.
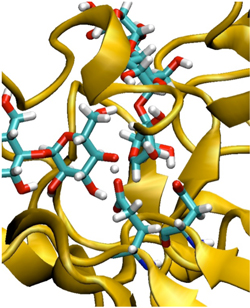
Figure 1. The transition state in the hydrolysys reaction of a polysaccharide subtrate in the a-amylase kinase.
- The reactivity analysis for a subtrate in the kinase reactions
- Understanding of the molecular function driven by the proton concentration gradient, which is the most important function for the biological energy conversion.
Nakamura HARUKI, Professor, Institute for Protein Research, Osaka University
The first principle calculation for all-electrons of the molecule such as protein by the molecular orbital (MO) method and DFT theory
MO, DFT theory, Hybrid DFT with Hartree-Fock
Quadrature of the multicentered Euler-Maclaurin-Lebedev, eigenvalue problem, and so on
MPI(to be extended to the hybrid parallelization)
Fortran77, Fortran90, MPI, HDF, ScaLAPACK, LAPACK, etc
Source code is available through ISLIM download site.
- Pigments like bacterio-chlorophyll
- Several-hundred to one-thousand-electrons system with 8,192 cores
- Required memory/disk storage: 1.2GB per core/100 MB
- The electron structure at the ground state and the excitation states for a several-hundred to several-thousands-of-electrons system with 640 thousands of cores
- Required memory/disk storage:1.6GB per core/several GB.
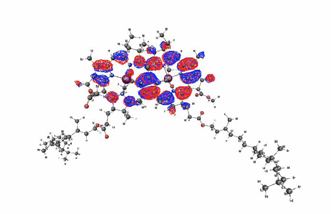
Figure 1. The homo orbital for the pigment (bacterio-chlorophyll dimer) at the photosynthetic reaction center derived from Rhodobacter sphaeroides.
- Understanding of the electron structure at the ground and excitation states for a protein system that contains transition metal
- Understanding of the reaction mechanism by the electron structure analysis
- The parameter calculations, such as the electromagnetic interaction, given a probe like the Electron Paramagnetic Resonance (EPR), for unknown protein structure.
Nakamura HARUKI, Professor, Institute for Protein Research, Osaka University
The code gives the precise analysis for a biological polymer such as a kinase reaction mechanism by introducing the electron state effect to the molecular mechanics (MM) approach
Molecular orbital method, molecular dynamics (MD) method
The direct solving method for dense matrix diagonalization, the iterative solving method for sparse-matrix simultaneous equations, an improved Wolf method, etc
Domain decomposition (MO), atom dividing (MD)
FORTRAN77, Fortran90, C, MPI, OpenMP, ScaLAPACK, LAPACK
To be released.
- The isomerization's thermal mechanical process for proline and N-metylacetamide (quantum system) in the water (classical treatment for H2O)
- Twenty thousands of atoms by a 256-core x86 cluster
- Required memory/disk storage: 300MB per core/3GB
- The reactive free-energy map for a several-hundred to several-thousands-of-electrons system surrounded by several hundred thousands of the classical atoms using 640 thousands of cores
- The dynamics analysis for both the ground and excitation states
- Required memory/disk storage: 1.6GB per core/several PB.
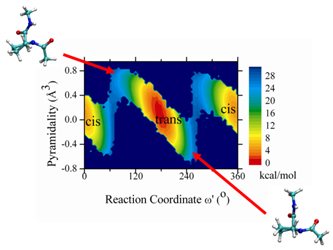
Figure 1. Coupled quantum-classical calculation for the reaction coordinate dependency of pyramidality.
- Useful knowledge for the kinase reaction process being important for life and the drug development controlling such reactions.