
 Strategic Programs for Innovative Research Field 1,
Strategic Programs for Innovative Research Field 1,
Supercomputational Life Science
Theme 2 Simulation Applicable to Drug Design
Innovative molecular dynamics drug
design by taking advantage of excellent
Japanese computer technology

Professor, Research Center for Advanced Science and Technology,
The University of Tokyo
Hideaki Fujitani
(Theme2 GL)
●Shooting for Real Drug Discovery
Thanks to the advent of the K computer, improvement in computer capacity enabled molecular dynamics calculation to see how a drug molecule acts on and binds to a disease target protein. With this step, an IT innovation in drug discovery, in which atomic-level changes in protein shape are revealed for drug design, is about to begin.
In Strategic Programs for Innovative Research Field 1 Supercomputational Life Science, Theme 2 Simulation Applicable to Drug Design which we are engaged in, we make the fullest possible use of the K computer to establish new Computer Aided Drug Design (CADD) technologies for innovating the drug discovery process, as well as to discover real drugs. Although they are both within the field of molecular simulation, Theme 1 deals with broader phenomena with biological importance. In Theme 2, objects are narrowed down to disease target proteins which are the target of drug discovery. This is the feature of Theme 2.
Until now, neither national institutes nor national universities have discovered drugs independently. This is because drug development including clinical trials is very expensive, costing 20 to 30 billion yen. In addition, in the step before clinical trials, most institutions and universities unfortunately have facilities that are not suitable to synthesize numerous drug candidate compounds. Since antibody drugs are proteins anyway, basically any university can synthesize them and does not have to rely on pharmaceutical companies for synthesis. However, synthesis of lowmolecular weight compounds costs a fortune for their design. Therefore, it is impossible at present for universities and institutes to do the entire process of drug discovery from the development phase to clinical trials.
For those reasons, we arrange tie-ups with pharmaceutical companies from the very beginning in our project, and proceed with the development in the form of collaborative study. We are obliged to get pharmaceutical companies involved to translate our project into real drug discovery. You may think government funds are being used for business, but the facts are contrary to that. In the research and development phase, companies cannot avoid expenses. In order to get them involved in collaborative studies even in this situation, we have to produce convincing simulation results. For discovering a real drug, both we and pharmaceutical companies have to take risks and make a serious effort.
●It is Important to Become a Pioneer of IT Drug Discovery
In these twenty years, drug discovery by computer simulation has been attempted many times but not yet fulfilled. The major reason for this is we did not have enough computation power to calculate the protein itself. In order to simulate a phenomenon in which a compound that may become a drug binds to a protein in solution and inhibits the function of the protein, at least 50 to 100 thousands of atoms are involved and 200 thousands of atoms are involved when the protein is large. Then about several million calculations are required. In addition, it was recently revealed that whether a compound is active is not is determined by simulating only one protein molecule, and simulation of the whole system is necessary. Then, the number of atoms involved easily surpasses 1 million. At last, the K computer has given us the computation power that we wanted. So far it has been determined empirically whether a compound binds to a target protein, but, now it becomes clear by computer-aided calculation. At last we have created environments for developing drugs logically.
The IT drug discovery (Fig. 1) efforts we are engaged also started in Europe and the US at almost the same time. You may know Mr. David E. Shaw (US) who made a special computer for MD calculation, ANTON, and is proceeding with IT drug discovery together with Mega Pharma (mega pharma company). Due to those activities, Japanese pharmaceutical companies which expressed their doubts became interested in IT drug discovery. However, since it has not yet produced any favorable results worldwide, they have not reached the stage for investing in and working on IT drug discovery. So we decided to become a pioneer in this field.
Now that we are on the starting line with other developed countries, it is important to start off as a pioneer. While collaborating with researchers from pharmaceutical companies in efforts geared toward real drug discovery and studying what to calculate, which result we should use and how to use the result in compound design, I believe we can expand the horizons of research and development of IT drug discovery. After they understand that they really can develop drugs through IT drug discovery, supercomputers comparable to the K computer will be introduced in major pharmaceutical companies possibly 5 years from now to promote further research. Opening up such a new epoch is the final goal of this project in a sense, and I also think it is one of the important missions for the K computer.
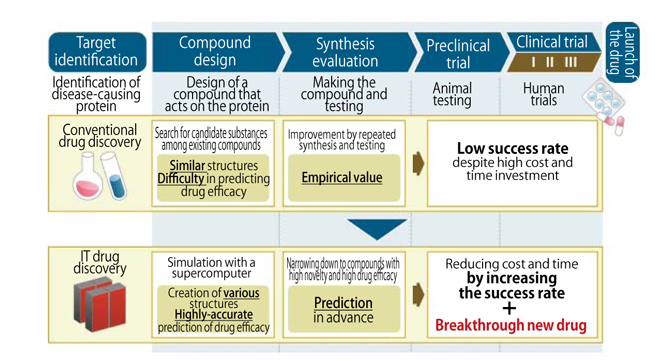
Fig.1 : Difference between conventional drug discovery and IT drug discovery
Figure by Fujitsu Limited
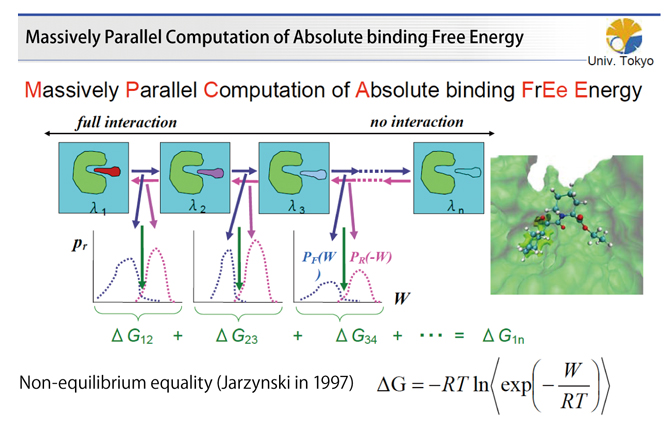
Fig.2 : Massively parallel computation of absolute binding free energy MP-CAFEE
In MP-CAFEE, massive molecular dynamics calculations are performed while changing drug-interaction parameters with proteins and a compound (drug) in a state of thermal agitation in solution. The binding free energy of the drug with the target protein is calculated from the work distribution when the parameter changes.
●High-accuracy Prediction of Drug Efficacy by Use of the High Computing Power of the K computer
Since many drugs target proteins, we have to find a compound that interacts strongly with the intravital target protein (ligand) to discover a more effective drug. We aim at rapid and efficient development of low molecular weight drugs in the following manner. By use of molecular dynamics calculations using the supercomputer, we run a simulation of a system including a target protein and a drug candidate compound, investigate protein-compound interaction, and design a new compound which acts only on the target protein.
For this purpose, we devised the MP-CAFEE method (Fig. 2) which is an algorithm for calculating free binding energy using the Jarzynski relation for free energy difference and non-equilibrium work discovered by Jarzynski in 1997. In this method, molecular dynamics calculations are performed on multiple intermediate states between two states. In one state, full interaction exists between the compound and other molecules. In the other virtual state, interaction disappears completely and the compound is uncoupled. Then, the free binding energy is calculated from the work necessary for the transition to the adjacent state. This enables accurate computation of the free binding energy between the target protein and the compound in solution. The feature of the MP-CAFEE method is that its accuracy is always high for any kind of protein and is not influenced by protein characteristics due to atomic-level calculation. Since such a huge calculation power is required for that, people often asked to us what kind of computer on earth we were going to use when we announced the project in 2005. However, thanks to the development of the K computer, conditions for producing tangible results are now satisfied.
In May 2011, we began work on porting Massively Parallel Computation of Absolute binding Free Energy (MP-CAFEE) into the K computer. So far, we have confirmed that MP-CAFEE works correctly in the K computer. Presently we are making adjustment for faster calculation. In August, we started calculations for actual development of drugs against target proteins of cancer and leukemia by using MP-CAFEE modified for the K computer. This requires searching for candidate compounds that may work well among several millions of compounds while checking their chemical structures. Therefore, we collaborate with the Bio-IT Business Development Unit, Fujistu Limited which runs a computer-based drug design business, and selected several hundreds of candidate compounds for MP-CAFEE calculations. Candidate compounds include not only existing compounds. We also design novel compounds that work better. In such a case, we of course check whether it can be synthesized and is free of toxicity (side effects). It is an advantage of IT drug discovery that it can reduce development costs and time. However, its maximum benefit is that we can design various new compounds and predict candidate compounds through molecular simulation without being limited by existing compounds.
In this fiscal year, I believe we can produce a series of drug candidate compounds against relatively small target proteins. Because of computation time, we give priority to the surest thing, and then prepare to handle larger and more difficult targets in the future. We will practically complete this project in three years. The biggest goal of our project is to develop several compounds that may be studied in clinical trials by then. Of course it includes the results we will produce in this fiscal year.
As I mentioned before, it is one of the objectives of this project to get Japanese pharmaceutical companies interested in IT drug discovery that has been failing to produce successful results, and to have them work with us. Thanks to worldwide interest and the completion of the K computer, we have received favorable responses and offers from pharmaceutical companies. Since it is a national project, we have talked with many pharmaceutical companies about collaboration. Many companies have already given us various proposals. Some of them wish to start during this fiscal year. Since we are not yet ready to get off the block, we are now continuing discussions with them and are preparing for the future.
BioSupercomputing Newsletter Vol.7
- SPECIAL INTERVIEW
- Interview with “K computer” Developer regarding Efforts in Exascale and Coming Supercomputer Strategies
Executive Architect, Technical Computing Solutions Unit, Fujitsu Limited
Motoi Okuda - Large-scale Virtual Library Optimized for Practical Use and Further Expansion into K computer
Professor, Department of Chemical System Engineering,
School of Engineering, The University of Tokyo
Kimito Funatsu
- Report on Research
- Old and new subjects considered through calculations of the dielectric permittivity of water
Institute for Protein Research, Osaka University
Haruki Nakamura
(Molecular Scale WG) - Development of Fluid-structure Interaction Analysis Program for Large-scale Parallel Computation
Advanced Center for Computing and Communication, RIKEN
Kazuyasu Sugiyama
(Organ and Body Scale WG) - SiGN : Large-Scale Gene Network Estimation Software with a Supercomputer
Graduate School of Information Science and Technology,
The University of Tokyo
Yoshinori Tamada
(Data Analysis Fusion WG) - ISLiM research and development source codes to open to the public
Computational Science Research Program, RIKEN
Eietsu Tamura
- SPECIAL INTERVIEW
- Understanding Biomolecular Dynamics under Cellular-Environments by Large-Scale Simulation using the “K computer”
Chief Scientist, Theoretical Molecular Science Laboratory,
RIKEN Advanced Science Institute
Yuji Sugita
(Theme1 GL) - Innovative molecular dynamics drug design by taking advantage of
excellent Japanese computer technology
Professor, Research Center for Advanced Science and Technology,
The University of Tokyo
Hideaki Fujitani
(Theme2 GL)
- Report
- Lecture on Computational Life Sciences for New undergraduate Students
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada