
 Strategic Programs for innovative Research Field 1
Strategic Programs for innovative Research Field 1
Supercomputational Life Science
Theme 1 Simulation of Intracelluar Molecular Dynamics
Understanding Biomolecular Dynamics
under Cellular-Environments by Large-Scale
Simulation using the“ K computer”

Chief Scientist, Theoretical Molecular Science Laboratory,
RIKEN Advanced Science Institute
Yuji Sugita
(Theme1 GL)
●Overview of the Project
Simulation studies in life science are now one of the most active research fields with rapidly progressing development of methodologies and algorithms. Many experimental data such as genome information, atomic structures and intracellular protein expression information are obtained one after another. Conventional life science focused on only the experimental data. However, it’s about time that we integrate these experimental data and promote better understanding of biological phenomena in a living system. In that sense, simulations achieved by the excellent computing capability of the “K computer” can be said to play a great role in changing life science into a new, predictable and controllable research system. It was in these circumstances that Strategic Programs for Innovative Research Field 1 Supercomputational Life Science began. In the theme we are engaged in (biomolecular simulations under cellular-environments), we aim at understanding and prediction of intracellular molecular dynamics by performing large-scale computer simulations on a molecular and cellular scale in which we focus intensively on the cellular environment. Many biomolecular simulations have been done so far. However, most of them tried to reveal the behavior of a single protein or DNA in aqueous solution or in a lipid bilayer membrane. When computed from the number of proteins in the cell and size of the cell, there is no doubt that the cytoplasmic environment is very different from the environment of aqueous solution. This has also been revealed experimentally. Just near one acting protein, there are other proteins. It is not yet completely understood how such an environment (intracellular molecular crowding environment) influences protein structure, stability and functions. In previous theoretical studies, the macromolecular crowding was studied using a very simple model in which a protein molecule was approximated to one particle. But, under this assumption, detailed intra-molecular interactions cannot be taken into account in the simulations. In this project, we would like to look at a lot of proteins under conditions close to the cellular environment on an unprecedented scale.
We have the following three sub-projects: The first is “Molecular and cellular-scale simulations in the cellular environments” for developing a new research field by cooperation between single-particle simulations and molecular dynamics simulations. The second is “Substance transport across the cell membrane by membrane proteins” for achieving a quantitative and predictive molecular simulation by free energy calculations based on extensive molecular dynamics simulations. And the third is “Understanding the dynamic structures and functions of DNA and proteins in a nucleus”, analyzed with an all-atom and coarse graining molecular dynamics calculation. For understanding life phenomena and linking them to prediction, it is essential to understand a “whole cell” by integrating molecular information with cellular information. For this purpose, at the end of the project, we would like to define the next challenge aiming at “Whole cell simulation” by using the findings obtained from research and development.
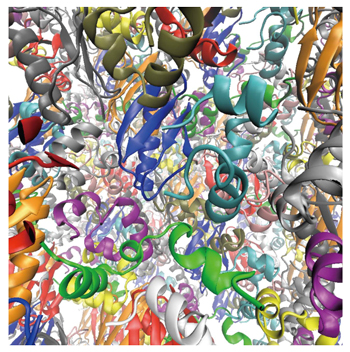
Understanding of the cellular environments and their role on biomolecules is the first step toward the whole cell modeling.
 |
 |
|
In order to see the molecular dynamics of an ion pump in biological membranes by simulation, a long stream of computing including about 260 thousand atoms such as proteins, lipid bilayer, water and ions, as well as their molecular interactions, is required. |
Simulation by changing protein concentration. Protein molecules crowded in cytoplasm are simulated by molecular simulation, showing the differences in hydration effect between in dilute condition and in crowded environment. |
●Goal of Molecular Simulation Studies
In order to realize Theme 1, “Molecular and cellular-scale simulations in the cellular environments,” two elements are necessary. One is simulation of the “considerably slow motion” of biomolecules such as proteins and nucleic acids. The other is elucidation of cellular functions from the molecular point of view by connecting molecular-scale (in the atomic detail) studies with cellular-scale (in the molecular details) studies. As for the former, we would like to attempt the simulation of millisecond-scale structural changes. Although I said it is slow, it is slow as our researchers’ standards. Actually it is an extremely fast molecular movement. However, in current molecular simulation studies, the time scale of long dynamics is micro-seconds. Therefore, millisecond molecular movement is a thousand times longer than microsecond molecular movement, and thus far slower. The computation environment available to us before completion of the “K computer” was about 100TFLOPS or so. Thanks to the advent of the “K computer”, it is now enhanced 100 times (the calculation performance of “K computer” is about 10PFLOPS (10,000TFLOPS)). It is actually considerably difficult to make a calculation 1,000 times longer by similar utilization of computers. Therefore, by developing novel computational approaches, we try to see millisecond dynamics. For this purpose, we have already been developing advanced parallelization techniques. For example, we have an algorithm called the multi-dimension replica-exchange molecular dynamics calculation. In this method, molecular dynamics calculations at different temperatures or with different parameters are performed in parallel for the copy of a system called a replica, and by exchanging temperature or another parameter at a certain frequency, the calculation can be accelerated. It is difficult to do parallelization with several ten thousand CPUs for a single molecular dynamics calculation. However, in the replica-exchange method, the molecular dynamics calculation of each replica is parallelized with several hundred to several thousand CPUs. By providing several ten to several hundred replicas, simultaneous and efficient utilization of several ten thousand CPUs is enabled. We have to integrate those various methods. Although we use the “K computer” having unprecedented computing performance, what we can do only by computation is limited as I have already described. Therefore, I believe collaboration with experiment is of the essence in order to implement this project. For example, simulations can collaborate with structural biology, in particular, X-ray crystallography and NMR (nuclear magnetic resonance). In addition, we can collaborate with single-molecular experiments to measure dynamics of proteins and protein complexes in cell in a most direct way. Experimental difficulties often arise from the time-resolution, whereas simulations usually suffer from insufficient sampling due to the short computational time. At milliseconds, real-time experimental measurements correspond with the simulation results at the same time scale.
●Results of Molecular Dynamics Simulations
While proceeding with development and upgrading of software for efficient utilization of the computing performance of the “K computer”, some simulation studies have already been implemented and produced results. One of them is the simulation, which takes account of the cytoplasmic molecular crowding environment, and we are taking the lead in implementing this simulation study. In the study, we could show the differences in hydration of proteins between in dilute condition and in crowded environment. This difference is essentially important for protein conformational stability, dynamics, and biochemical functions. Since microscopic parameters for the intracellular environment are difficult to measure experimentally, this computational result can be said to be quite helpful also for experimentalists. By utilizing the “K computer” hereon, molecular crowding analysis in much large-scale systems will be studied. Further progress in this research is expected. Molecular simulation of membrane transport protein has also made great strides. Conventionally, it took some time to provide conformational dynamics of membrane proteins by molecular simulation after clarification of X-ray crystal structure. Recently, we can perform molecular dynamics calculations including a lipid bilayer membrane immediately after the determination of the X-ray crystal structure. Structural change taking place partly in the transport cycle of membrane transport proteins has already been unveiled by simulations and experiments. We expect to perform molecular dynamics simulations to reveal substance transportation across membranes by membrane transport proteins using the full power of the “K computer”. The project we are engaged in is expected to contribute not only to basic science, but also drug discovery and medical care. The information observed here would be useful for such applications in near future. Finally, I would like to emphasize again the importance in the developments of simulation methodologies and models to achieve our research goals. We also need strong computational skills to realize large-scale molecular and cellular simulations in this project. For this, the participation of young researchers or students is requisite. To increase the number of those people is also one of the tasks of the project over the long term.
BioSupercomputing Newsletter Vol.7
- SPECIAL INTERVIEW
- Interview with “K computer” Developer regarding Efforts in Exascale and Coming Supercomputer Strategies
Executive Architect, Technical Computing Solutions Unit, Fujitsu Limited
Motoi Okuda - Large-scale Virtual Library Optimized for Practical Use and Further Expansion into K computer
Professor, Department of Chemical System Engineering,
School of Engineering, The University of Tokyo
Kimito Funatsu
- Report on Research
- Old and new subjects considered through calculations of the dielectric permittivity of water
Institute for Protein Research, Osaka University
Haruki Nakamura
(Molecular Scale WG) - Development of Fluid-structure Interaction Analysis Program for Large-scale Parallel Computation
Advanced Center for Computing and Communication, RIKEN
Kazuyasu Sugiyama
(Organ and Body Scale WG) - SiGN : Large-Scale Gene Network Estimation Software with a Supercomputer
Graduate School of Information Science and Technology,
The University of Tokyo
Yoshinori Tamada
(Data Analysis Fusion WG) - ISLiM research and development source codes to open to the public
Computational Science Research Program, RIKEN
Eietsu Tamura
- SPECIAL INTERVIEW
- Understanding Biomolecular Dynamics under Cellular-Environments by Large-Scale Simulation using the “K computer”
Chief Scientist, Theoretical Molecular Science Laboratory,
RIKEN Advanced Science Institute
Yuji Sugita
(Theme1 GL) - Innovative molecular dynamics drug design by taking advantage of
excellent Japanese computer technology
Professor, Research Center for Advanced Science and Technology,
The University of Tokyo
Hideaki Fujitani
(Theme2 GL)
- Report
- Lecture on Computational Life Sciences for New undergraduate Students
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada