

SiGN : Large-Scale Gene Network
Estimation Software with a Supercomputer

Graduate School of Information Science and Technology,
The University of Tokyo
Yoshinori Tamada
(Data Analysis Fusion WG)
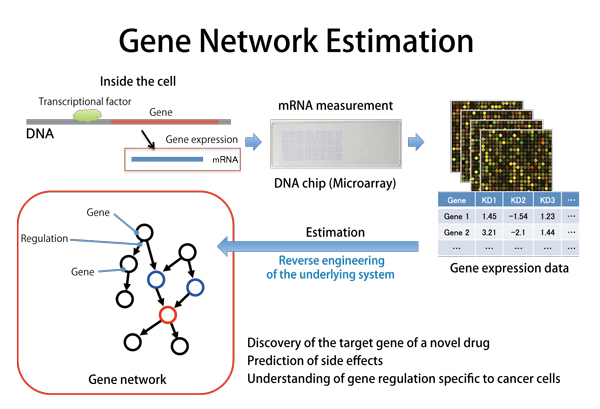
Human cells are said to have about 20 to 30 thousand genes. The human body is mostly composed of proteins. The gene is a blueprint for proteins made in the cell. As well as the kind of protein, the timing and quantity of protein production is also regulated by special genes. Those genes (≒ proteins) are also regulated by another gene. Briefly, genes form a complicated regulation network. Most of the system is still poorly understood. Even among the same human cells, they have different networks in different organs. Drugs modify gene networks and cancer cells have destroyed networks. Gene network estimation is an approach to infer or estimate such gene regulatory networks (=gene network) from measurable data by mathematical, statistical and informational scientific methods. Although it is impossible to measure all proteins produced in the cell with current technologies, the amount of mRNA synthesized prior to protein production is measurable for every gene. Data measured like this is called gene expression data. Data obtained from one measurement are a snapshot of cell status. It is impossible to infer or estimate regulations between genes only with this one measurement. Massive data are necessary for that. Therefore, we collect data necessary for estimation by applying various stimuli to the cell, collecting cells from patients with a particular disease or taking data temporally at regular time intervals. Inference and estimation of gene networks enables clarification of regulations between genes by exhaustive computation instead of the conventional time-consuming method of searching for genes one by one and repeating experiments. It is expected that this approach will enable efficient development of new drugs, identification of cancer-specific genes, and understanding of the functions of such genes.
SiGN is software for estimating a gene network with a supercomputer from gene expression data. As the gene network, various models have been proposed. However, every model has both merits and demerits. None of them is by far the best. After deciding a model, we still have to choose a method for estimating parameters from the data. Those methods also have good and bad points. SiGN is a gene network estimation software implementing multiple gene network models and estimation algorithms, both of which requires vast amount of computation time assuming computation using a supercomputer. In particular, SiGN is composed of three sub-programs, SiGN-BN using static and dynamic Bayesian networks, SiGN-SSM using a State Space Model (SSM) and SiGN-L1 implementing a parameter estimation method by L1 regularization. SiGN-BN implements a new algorithm called NNSR. Conventionally, gene network estimation using Bayesian networks was applicable to about 1000 genes. Now it is applicable to all genomes (all genes) thanks to NNSR. Temporal data allows SiGN-SSM to estimate dynamic gene networks that are able to be simulated. It does not give the network structure but the strength of relationships among all genes as mathematical values. Thanks to supercomputers, network structures which have been difficult to compute, are now computable with a degree of confidence. L1 regularization was originally applicable to large-scale gene networks. However, the computation time of conventional methods is not enough to estimate networks focusing on individual differences in gene expression. By using the K computer, it is able to be computed within a realistic time-frame.
Development of SiGN is targeted mainly at the K computer and Shirokane, a supercomputer of the Human Genome Center. Several sub-programs have already been installed in Shirokane and are available for users. For more information, please contact the SiGN website at http://sign.hgc.jp.
 |
BioSupercomputing Newsletter Vol.7
- SPECIAL INTERVIEW
- Interview with “K computer” Developer regarding Efforts in Exascale and Coming Supercomputer Strategies
Executive Architect, Technical Computing Solutions Unit, Fujitsu Limited
Motoi Okuda - Large-scale Virtual Library Optimized for Practical Use and Further Expansion into K computer
Professor, Department of Chemical System Engineering,
School of Engineering, The University of Tokyo
Kimito Funatsu
- Report on Research
- Old and new subjects considered through calculations of the dielectric permittivity of water
Institute for Protein Research, Osaka University
Haruki Nakamura
(Molecular Scale WG) - Development of Fluid-structure Interaction Analysis Program for Large-scale Parallel Computation
Advanced Center for Computing and Communication, RIKEN
Kazuyasu Sugiyama
(Organ and Body Scale WG) - SiGN : Large-Scale Gene Network Estimation Software with a Supercomputer
Graduate School of Information Science and Technology,
The University of Tokyo
Yoshinori Tamada
(Data Analysis Fusion WG) - ISLiM research and development source codes to open to the public
Computational Science Research Program, RIKEN
Eietsu Tamura
- SPECIAL INTERVIEW
- Understanding Biomolecular Dynamics under Cellular-Environments by Large-Scale Simulation using the “K computer”
Chief Scientist, Theoretical Molecular Science Laboratory,
RIKEN Advanced Science Institute
Yuji Sugita
(Theme1 GL) - Innovative molecular dynamics drug design by taking advantage of
excellent Japanese computer technology
Professor, Research Center for Advanced Science and Technology,
The University of Tokyo
Hideaki Fujitani
(Theme2 GL)
- Report
- Lecture on Computational Life Sciences for New undergraduate Students
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada