

Biosupercomputing Pioneers the Future of Life Science
Large-scale Virtual Library Optimized for Practical Use and Further Expansion into K computer

Professor, Department of Chemical System Engineering,
School of Engineering, The University of Tokyo
Kimito Funatsu
●Present status of compound library, a key for drug discovery
New drug development is very time-consuming, taking a dozen years or so. It is also said that only one out of several ten thousand compounds is sent to the medical work front. Therefore, it is a reality that both the development cost and R&D risk are extremely high. Such a new drug development starts from identification of a drug target, which is followed by discovery of a lead compound and its optimization for good activity. Then, the compound is subjected to clinical trial. The key point for success is the on-target capture of a group of lead compounds in the early stages which starts with screening of a compound library for a lead compound. This is why the potential development capability of a pharmaceutical company is dictated by the chemical variety, quality and scale of the compound library it owns.
Then what about the current status of compound libraries? It is estimated that the theoretical total of compounds which may become drug discovery targets is 10 to the sixth power. Meanwhile, existing compound libraries owned by pharmaceutical companies (megapharma) cover only several million compounds. This results in unsuccessful screening without a hit, or discovery of only low-active compounds. The large number of absent compounds has always been a major concern. For those reasons as well as improvement in the hit ratio of screening, an increase in the scale and variety of the compound library is strongly desired. From a viewpoint of searching more promising compounds exhaustively, we expect much from the use of virtual libraries constructed in computers. However, existing virtual libraries stock only several 10 million compounds at most. They are paltry compared to the theoretical total. You can get only a partial view of chemical space from their screening, so the fact is they are not very helpful. Since it is a virtual library, it has another problem. Even if we can make a short list of a group of high-score lead compounds, study of their synthesis would require great cost. If we rely only on theoretical manipulation based on a combination of atomic species and bond orders each atom can have for creation of a virtual compound structure, the long-awaited library may include many unstable compounds which cannot be synthesized.
●Feature and brief overview of large-scale virtual library
“The large-scale virtual library composed of chemical structures able to be synthesized and reaction schemes” we are engaged in constructing, is an unprecedented new virtual library that solves the aforementioned problems and develops an acceptable quality, variety and scale of the group of virtual compounds. We don’t intend to simply make a “largescale virtual library”. It is important that the library consists of “chemical structures able to be synthesized and reaction schemes”. The library also includes synthesis routes for producing compounds. Of course, scale is also of the essence. We aim at construction of a 1 to 2 billion-scale virtual library, which exceeds the existing tens of millions-scale library, while ensuring drug-likeliness and variety. What we have developed for this purpose is a system to create new structures, in which seed structures are put into the construction system sequentially from the existing compound library covering 420 thousands compounds, followed by application of structure transformation information called Transform. Transform is structural change information on the reaction site before and after reaction, which is extracted from a reaction database. In short, Transform is a database built by extracting and storing information on changes in bond orders and structural environments at reaction sites of the reactant and the product of each reaction scheme, and or “essence of the reaction” from existing reaction databases.
The virtual library with ordinary synthesis routes is constructed by applying Transform information and continuously running an Ordinary Synthesis Reaction Construction System that presents product structure for reactant structure as a reaction scheme. Compound structures included in the virtual library form an ordinary synthesis tree structure that maintains a relation between reactant structure and product structure. In contrast, by continuously running a Retrosynthesis Reaction Construction System that presents a precursor structure for the target compound structure as a reaction scheme, a virtual library with retrosynthesis routes is constructed. Compound structures included in this virtual library form a retrosynthesis tree structure that maintains a relation between synthetic precursor structure and product structure. Schemes for predicting products from reactants shown in the ordinary synthesis tree domain, and a synthetic precursor with the reaction site, are proposed from the target structure in the retrosynthesis tree domain. Briefly, virtual compound structures not only from the ordinary reaction direction but also from the retrosynthesis direction suggesting the starting material, are included in the virtual library.
When a seed structure of the existing compound library is actually entered, several candidates for the product structure appear by applying Transform. Then by applying Transform to the candidates for the product structure as a reactant structure, candidates for the next-level product structure come out. On the other hand, you can track back to the starting material for a certain compound. Some compounds have small molecular size. Or they lack drug-likeliness and are considered to be inadequate as candidates for the lead compound. They are not retrieval objects of the virtual library. However, since they are necessary information in the sense of connecting synthesis routes, they are included in the library.
In this fiscal year, we are continuing scale expansion by converting the output structure into an input structure recursively and generating a multistage scheme, and aim at storing a total of 1 billion not-overlapping, unique compounds including both ordinary- and retro-reaction schemes in the virtual library as a whole. By making them branch further and adding initial seed structures, I believe a virtual library with 2 billion compounds would be attainable.
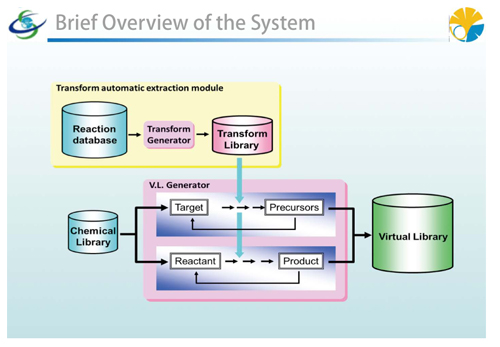 |
Brief overview of the large-scale virtual library system |
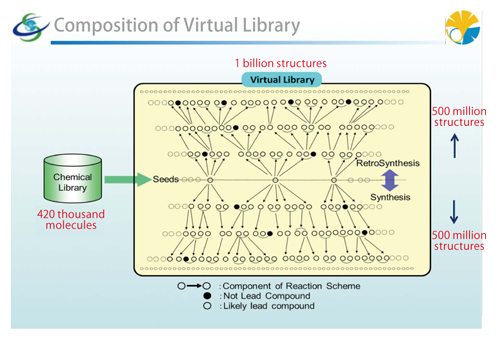 |
Composition of the large-scale virtual library |
●Assessment of library construction engine
While continuing further development, we make assessments for understanding the features of the virtual library construction engine and groups of output compounds. Although all compounds are not covered, when we generated 15 million compounds from 420 thousand structures used as a group of seed structures, we got 6.3 million non-overlapping, unique chemical structures. The overlapping percentage of output compounds was 58%, a little more than half. As regards novelty, when the group of 6.3 million generated compounds was compared with the existing compound library including 15 million available compounds, the overlapping percentage with commercially-available compounds was only 1.33%. Therefore, most structures output by this system were novel compounds. I conclude that novelty is fully ensured.
We are also examining the influence of character distribution of the group of input compounds. The virtual library is generated from the input seed structures. As criteria to determine whether the generated chemical structure has significance as a drug, we have ADMIT (absorption, distribution, metabolism, elimination and toxicity) characteristics. By calculating the characteristic prediction, we can assess the chemical structure preliminarily. For example, an orally administered compound does not work as drug if it is not absorbed into the body. Drugs are organic compounds and have considerably large molecular weights, so basically they are insoluble in water. Naturally, they are absorbed poorly. Since this is no good to us, those compounds need to maintain a certain level of water solubility. Solubility is not a sole determinant. A large polar surface area may increase solubility. Or, more hydrogen bond donors or receptors may help them blend with water. Like this, they can be assessed not only by solubility but also other characteristics. The empirical rule for predicting such absorption is “Lipinski's rule of five”. This time, we examined the distribution of each characteristic value shown by “Lipinski's rule”. As a result, we confirmed that the virtual chemical structures output by this system take over characteristics of the group of input compounds while expanding the distribution of characteristics values. We also confirmed that when using a group of compounds with high conformance ratio for each characteristic value index as a seed, the system can output a group of chemicals with high index conformance ratio. Briefly, it shows that appropriate selection of the group of seed structures enables output of virtual compounds suitable for drugs with high probability.
After this, we will actually input this large-scale virtual library to the “K computer” and have it used by users including the public. Since screening software is developed by another group, we provide the large-scale virtual library. If a library construction engine for constructing elemental chemical structures and reaction schemes is put into the “K computer”, pharmaceutical companies, would-be users of the “K computer”, would be able to construct a virtual library from their own compound library. The library construction engine itself has already been developed by Funatsu Laboratory. Since there is a great demand for this engine, we think we will be providing this construction engine.
We think we have almost finished preparations. From now on, we have to think how to use the large-scale virtual library for promoting its practical use toward actual drug discovery while listening to what users want. I can say that the large-scale virtual library is entering a new phase.
 |
|
BioSupercomputing Newsletter Vol.7
- SPECIAL INTERVIEW
- Interview with “K computer” Developer regarding Efforts in Exascale and Coming Supercomputer Strategies
Executive Architect, Technical Computing Solutions Unit, Fujitsu Limited
Motoi Okuda - Large-scale Virtual Library Optimized for Practical Use and Further Expansion into K computer
Professor, Department of Chemical System Engineering,
School of Engineering, The University of Tokyo
Kimito Funatsu
- Report on Research
- Old and new subjects considered through calculations of the dielectric permittivity of water
Institute for Protein Research, Osaka University
Haruki Nakamura
(Molecular Scale WG) - Development of Fluid-structure Interaction Analysis Program for Large-scale Parallel Computation
Advanced Center for Computing and Communication, RIKEN
Kazuyasu Sugiyama
(Organ and Body Scale WG) - SiGN : Large-Scale Gene Network Estimation Software with a Supercomputer
Graduate School of Information Science and Technology,
The University of Tokyo
Yoshinori Tamada
(Data Analysis Fusion WG) - ISLiM research and development source codes to open to the public
Computational Science Research Program, RIKEN
Eietsu Tamura
- SPECIAL INTERVIEW
- Understanding Biomolecular Dynamics under Cellular-Environments by Large-Scale Simulation using the “K computer”
Chief Scientist, Theoretical Molecular Science Laboratory,
RIKEN Advanced Science Institute
Yuji Sugita
(Theme1 GL) - Innovative molecular dynamics drug design by taking advantage of
excellent Japanese computer technology
Professor, Research Center for Advanced Science and Technology,
The University of Tokyo
Hideaki Fujitani
(Theme2 GL)
- Report
- Lecture on Computational Life Sciences for New undergraduate Students
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada