

Free Energy Profile Calculations for Changes
in Nucleosome Positioning with All-Atom
Model Simulations
HPCI Strategic Program Field 1 Supercomputational Life Science
Quantum Beam Science Directorate, Japan Atomic Energy Agency
(From left) Hidetoshi Kono, Hisashi Ishida, Yoshiteru Yonetani, Kimiyoshi Ikebe
(field1- Program1)




Human genes are encoded by a genome DNA which consists of approximately 3billion base pairs and approximately 2meters long. The genome DNA of eukaryotes including that of humans, forms chromatin structures and is stored compactly in the cell nucleus whose diameter is approximately severalμmeters long. The fundamental, repeated structure unit of chromatin is called nucleosome, in which about 150 base-pair DNA is wrapped nearly twice around a protein core called a histone. Recently it has been found that structural changes in the genomic DNA play key roles in the regulation of gene expression. While our genes are protected from being damaged and are stabilized by forming the nucleosome structure, during DNA metabolism which is the basis of vital functions such as transcription, duplication, repair and recombination, disruption of the nucleosome structure and changes in nucleosome positioning occur so that the DNA can directly interplay with regulatory proteins and RNA polymerases (Fig.1). The nucleosome structure has such seemingly-contradictory functions. Our interest is understanding the mechanism which brings about the disruption of the nucleosome structure and changes in nucleosome positioning.
In addition, recent studies have revealed that the changes in nucleosome structure affect the cell differentiation mechanism. The individual cells that constitute our body essentially have the same genetic information (genome DNA). However, once a cell becomes diferentiated, it usually only generates cells of the same type; a skin cell generates only skin cells and a liver cell only liver cells. Furthermore, recent research has revealed that the mechanisms of DNA metabolism and the cell differentiation are closely related to chemical modification of histone proteins that are a constituent of the nucleosome structure. In this way, changes in gene expression or cellular phenotype can be inherited as a kind of memory without accompanying any sequence change in the genome DNA, which is called epigenetics. This is one of the most active research areas in molecular biology nowadays.
In our study, we elucidate the mechanism of changes in nucleosome positioning from the aspect of the free energy profile by use of computer simulation. By examining how the difference in DNA base sequence, chemical modification of DNA, histone proteins and their variants affect the free energy profile, we pursue research on epigenetics. We have developed a computer program called SCUBA that is suitable for large-scale molecular dynamics simulations. We are among the first to introduce an algorithm in SCUBA which can divide the target system in space and enables us to efficiently compute it in parallel. With SCUBA, we have executed a dynamics analysis of Holliday Junction branch migration, which is observed in DNA homologous recombination, and a ribosome molecule in a system composed of over 2million atoms. As shown in Fig.2, it is known that nucleosomes spontaneously undergo slow conformational fluctuations whereby a partial unwrapping of DNA which was wrapped around the histone protein (opening) occurs due to thermal fluctuation, as if they were breathing. In our project, by using "K computer" we are aiming to clarify how such a dynamics and structural stability of nucleosomes are changed by chemical modification of the DNA and the histone proteins, compositional changes in histone variants, and differences in the base sequence of wrapped DNA. Since such a dynamics occurs in the order of sub-seconds, even the world’s fastest "K computer" cannot simulate for long enough to track the dynamics by simply waiting for thermal fluctuation phenomena to occur during computation. Therefore, we set an appropriate reaction coordinate, and induce a structural change along the coordinate to compute the free energy profile for the structural change. Through this molecular simulation approach, we intend to physicchemically understand the basic molecular mechanism of living processes, or how gene expression is regulated.
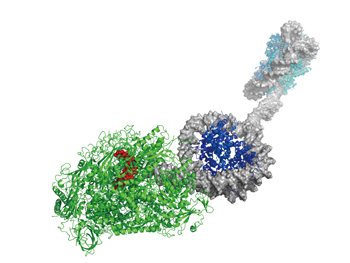 |
Fig 1 : Structural view of mRNA synthesis by RNA polymerase. How does RNA polymerase carry out transcription process against genome DNA that forms a nucleosome structure? We approach the problem through molecular simulation. |
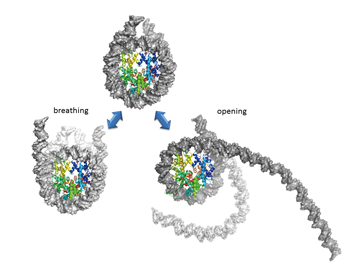 |
Fig 2 : We perform an analysis of the free energy profile for the structural change of nucleosomes shown in the figure through molecular simulation. It is experimentally suggested that partial unwrapping and rewrapping of DNA occurs spontaneously as if it were breathing. |
BioSupercomputing Newsletter Vol.6
- SPECIAL INTERVIEW
- Development of New Fluid-structure Interaction Analysis (ZZ-EFSI) Resulting in Rapid Achievement of High Operation Performance
Research Associate Professor, School of Engineering, The University of Tokyo Kazuyasu Sugiyama - Interview with High-performance Computing Team Members: Continued Efforts in Tuning to Harness the Potentials and the High Capability of the K Computer
Group Director of Computational Molecular Design Group,
Quantitative Biology Center, RIKEN
Makoto Taiji
Senior Researcher of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Yousuke Ohno
Senior Researcher of High Performance Computing Development Team,
High Performance Computing Development Group,
RIKEN HPCI Program for Computational Life Sciences
Hiroshi Koyama
Researcher of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Gen Masumoto
Research Associate of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Aki Hasegawa
- Report on Research
- Functional Analysis of Multidrug Efflux Transporter AcrB by All-Atom Molecular Dynamics Simulation
Graduate School of Nanobioscience,
Yokohama City University
Tsutomu Yamane,
Mitsunori Ikeguchi
(Molecular Scale WG) - Multi-scale Modeling of the Human Cardiovascular System
Computational Science Research Program,
RIKEN Liang Fuyou (Organ and Body Scale WG) - Toward a spiking neuron-level model of the early saccade visuomotor system
Kyoto University Jan Moren
Nara Institute of Science and Technology Tomohiro Shibata
Okinawa Institute of Science and Technology Kenji Doya
(Brain and Neural Systems WG) - Developing the MD Core Program for Large Scale Parallel Computing
Computational Science Research Program,
RIKEN Yousuke Ohno (High-performance Computing Team)
- SPECIAL INTERVIEW
- Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Program Director of RIKEN HPCI Program for Computational Life Sciences
Toshio Yanagida
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences
Akinori Kidera
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences
Yukihiro Eguchi
- Report on Research
- Free Energy Profile Calculations for Changes in Nucleosome Positioning with All-Atom Model Simulations
Quantum Beam Science Directorate, Japan Atomic Energy Agency
Hidetoshi Kono, Hisashi Ishida, Yoshiteru Yonetani, Kimiyoshi Ikebe (Field 1- Program 1) - Estimation of Skeletal Muscle Activity and Neural Model of Spinal Cord Reflex
Information Science and Technology, The University of Tokyo
Yoshihiko Nakamura (Field1 - Program 3)
- Report
- ISLiM Interim Accomplishment Meeting in 2011
Computational Science Research Program, RIKEN Eietsu Tamura - Computational Life Sciences Classes Held in High Schools
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada, Yasuhiro Fujihara, Yukihiro Eguchi