

Functional Analysis of Multidrug Efflux
Transporter AcrB by All-Atom Molecular
Dynamics Simulation


Graduate School of Nanobioscience, Yokohama City University
Tsutomu Yamane (left photo)
Mitsunori Ikeguchi (right photo)
(Molecular Scale WG)
Drug resistance is a phenomenon where drugs become ineffective against pathogens or cancer cells, which leads to a major social problem. There are some known mechanisms for drug resistance. One of the causes is where membrane proteins, located in the cell membrane of the bacteria and called multidrug efflux transporters, play an important role. A multidrug efflux transporter is a membrane protein which works to discharge various cytotoxic chemicals actively to the outside of the cell.
Among RND-type multidrug efflux transporters typically found in multidrug resistant Pseudomonas aeruginosa which is the main cause of nosocomial infection, the atomic-level crystalline structure of Escherichia coli-derived AcrB was determined in 2006 by Dr. Satoshi Murakami (presently professor of Tokyo Institute of Technology), et al. According to the results, AcrB functions as an assembly of three giant proteins (homotrimer), each of which consists of approximately 1,000 amino acids. The respective proteins have three different 3-D structures that act for access, binding and extrusion of the drug, respectively (Fig. 1A). With the drive force obtained by transfer of protons into cells using the difference in proton concentration (pH) of periplasm and cytoplasm, each protein switches between the 3-D structures in turn to perform the drug-efflux function (functionally rotating mechanism, Fig. 1B). In addition, three charged amino acids located at the membrane spanning region (Asp407, Asp408, Lys940) contribute to proton transfer into cells, and the side chain structure of Lys940 is different only in the extrusion state (Fig. 1C). Through this, a change in the protonated state of these charged amino acids due to proton transfer is believed to trigger the functionally rotating mechanism.
AcrB is the common res earch t arget of our Molecular Scale Team, and its molecular simulation has been performed by various methods. Since a more realistic environment is considered in the allatom molecular dynamics simulation we use, our computation covers a system including membrane lipids, water molecules, ions and even hydrogen atoms (Fig. 2, center). Therefore, our computations have to be performed on a very large system of which the total particle number is approximately 470,000, but we can take detailed views of the various interactions in the system. Such an all-atom molecular dynamics simulation revealed the following.
(1) |
When Asp407 and Asp408 were both in the deprotonated state in the extrusion protomer, it was found that the side chain of Lys940 changes to have the structure found in the binding state and the access state (Fig. 2, right panel). Meanwhile, when only Asp408 is in the protonated state, the extrusion-type conformation was found to be stably maintained. |
(2) |
In the simulation where both Asp407 and Asp408 of the extrusion protomer were deprotonated concurrently to change the struc ture of the Lys940 side chain, the drug entry point which is usually closed in the extrusion state opens, and a structural change toward the access state was observed (Fig. 2, left panel). |
Currently we are studying to reveal the mechanisms of how a structural change of the extrusion-type Lys940 side chain leads to a structural change for opening the drug entry point.
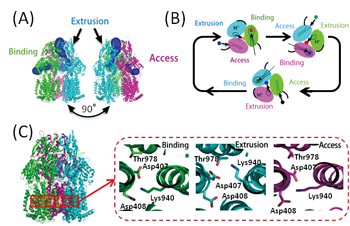 |
Fig.1 :Structure and drug efflux mechanism of multidrug efflux transporter AcrB |
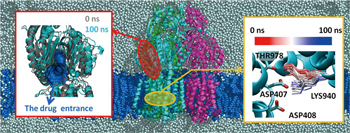 |
Fig.2 : System used for all-atom molecular simulation (center: a water molecule is indicated by a gray sphere, and a lipid by a blue sphere in the figure), and the results (left and right panels) |
BioSupercomputing Newsletter Vol.6
- SPECIAL INTERVIEW
- Development of New Fluid-structure Interaction Analysis (ZZ-EFSI) Resulting in Rapid Achievement of High Operation Performance
Research Associate Professor, School of Engineering, The University of Tokyo Kazuyasu Sugiyama - Interview with High-performance Computing Team Members: Continued Efforts in Tuning to Harness the Potentials and the High Capability of the K Computer
Group Director of Computational Molecular Design Group,
Quantitative Biology Center, RIKEN
Makoto Taiji
Senior Researcher of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Yousuke Ohno
Senior Researcher of High Performance Computing Development Team,
High Performance Computing Development Group,
RIKEN HPCI Program for Computational Life Sciences
Hiroshi Koyama
Researcher of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Gen Masumoto
Research Associate of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Aki Hasegawa
- Report on Research
- Functional Analysis of Multidrug Efflux Transporter AcrB by All-Atom Molecular Dynamics Simulation
Graduate School of Nanobioscience,
Yokohama City University
Tsutomu Yamane,
Mitsunori Ikeguchi
(Molecular Scale WG) - Multi-scale Modeling of the Human Cardiovascular System
Computational Science Research Program,
RIKEN Liang Fuyou (Organ and Body Scale WG) - Toward a spiking neuron-level model of the early saccade visuomotor system
Kyoto University Jan Moren
Nara Institute of Science and Technology Tomohiro Shibata
Okinawa Institute of Science and Technology Kenji Doya
(Brain and Neural Systems WG) - Developing the MD Core Program for Large Scale Parallel Computing
Computational Science Research Program,
RIKEN Yousuke Ohno (High-performance Computing Team)
- SPECIAL INTERVIEW
- Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Program Director of RIKEN HPCI Program for Computational Life Sciences
Toshio Yanagida
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences
Akinori Kidera
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences
Yukihiro Eguchi
- Report on Research
- Free Energy Profile Calculations for Changes in Nucleosome Positioning with All-Atom Model Simulations
Quantum Beam Science Directorate, Japan Atomic Energy Agency
Hidetoshi Kono, Hisashi Ishida, Yoshiteru Yonetani, Kimiyoshi Ikebe (Field 1- Program 1) - Estimation of Skeletal Muscle Activity and Neural Model of Spinal Cord Reflex
Information Science and Technology, The University of Tokyo
Yoshihiko Nakamura (Field1 - Program 3)
- Report
- ISLiM Interim Accomplishment Meeting in 2011
Computational Science Research Program, RIKEN Eietsu Tamura - Computational Life Sciences Classes Held in High Schools
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada, Yasuhiro Fujihara, Yukihiro Eguchi