

Developing the MD Core Program
for Large Scale Parallel Computing

Computational Science Research Program, RIKEN
Yousuke Ohno
(High-performance Computing Team)
Our team has been developing a MD core program for large-scale parallel computing to deliver high-speed core program libraries designed especially for a "K computer", and to understand and develop high-speed technologies for applications using "K computer ".
Molecular Dynamics (MD) simulation is a method for representing molecular activities and changes in structure by calculating the effects and forces acting on the atoms that make up molecules.
In life sciences, this is used to explain the nature of the biomolecule such as protein, and is the foundation of living phenomena. In biomolecule simulations for proteins, covalent bonding, Van der Waals, and Coulombs forces act on the atoms. For covalent bonding, there is a simple classical force field that acts like a harmonic oscillator, in which case sequences of up to four connected atoms are used, and the computational effort is proportional to the number of atoms in the covalent bond. But for Van der Waals and Coulombs, forces act between any atoms, meaning an interaction could exist between every pair of atoms. The computational effort in this case will be proportional to the number of atoms squared. The calculation effort for Van der Waals and Coulomb forces will take up most of the processing time if there are many atoms. The simplest way for speeding up the calculation is to ignore the forces between distant atoms and cut off calculation. Van der Waals forces decrease in inverse proportion to the power of 7 and 13 of the distance between atoms, and ignoring distances of 1.4nm and more will hardly affect calculation precision. Coulomb forces also decrease in inverse proportion, but with the power of 2, and a simple cut-off calculation will affect calculation precision. Therefore, a fast and common way for calculating Coulomb forces between distant atoms is the PME (Particle Mesh Ewald) method which uses FFT. However, FFT requires a large range of communication, and leads to a rise in communication time if the level of parallels is high. Therefore, we are considering using a calculation method that does not use FFT such as the fast multipole method (FFM).
As a large-scale test on cut-off calculation, we performed simulations on how "molecular crowding" affects protein dynamics. In an intracellular environment, proteins exist in a higher density than normal experiments, and it has been verified that the crowding of proteins affects their structure and interaction. To simulate protein crowding, it is necessary to perform simulations of a large number of proteins. Therefore, we have high hopes for "K computer" which is capable of large-scale computation. In a calculation test, we compared the structure of the TTHA1718 protein which is made of approximately 1,000 atoms. This was performed in an underwater environment, and in an environment of egg whites which are 30% ovalbumin proteins (consist of nearly 6,000 atoms). Fig.1 shows the fluctuation in positions of the amino acids that make up TTHA. Although we have not retested due to shortage of calculation time, we were able to confirm differences in TTHA activity in an underwater environment (red line vit1, blue line vit2), and an environment with ovalbumin (green line viv). Fig. 2 is a visualization of TTHA surrounded by ovalbumin.
In the performance evaluation of "K computer" (note), we were able to obtain an efficiency increase of 30%, an actual calculation speed of 1.3 PFlops using 1.8 hundred million atoms, and a cut-off calculation of 2.8nm. An efficiency increase of 40% or more has been achieved for the cut-off calculation part.
As of this point, we have implemented a cut-off calculation, the PME method, and FMM. Detailed algorithms of the PME method and FMMs have been refined for optimization. We have implemented a communication method that is designed for "K computer"'s mesh or torus network structure, but in cases where the number of atoms per node is small, the communication time exceeds the calculation time. Therefore, we are continuing improvements for reducing communication time.
Our research is a joint research project with the High-performance Computing Team whose members are Hiroshi Koyama, Gen Masumoto, Aki Hasegawa, Gentaro Morimoto, Noriaki Okimoto, Hidenori Hirano, and Naoyuki Miyashita from the Life System Research Center.
Some of the results of this research have been collected by using the RIEKN Integrated Cluster of Clusters (RICC) system.
(Note) Performance evaluation of a next-generation supercomputer, the petascale computer "K computer", by RIKEN's next-generation supercomputer development implementation division
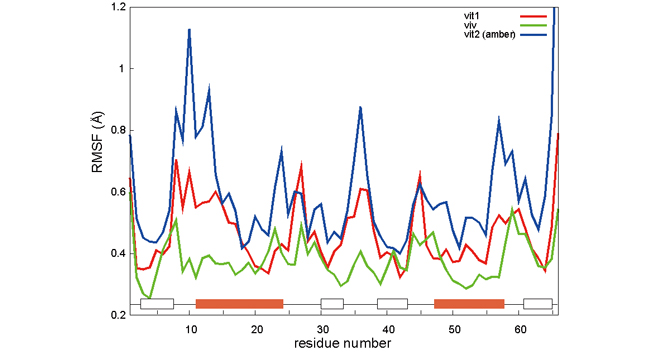
Fig 1 : Fluctuation in TTHA structure. Red and blue are underwater environments, and green is an environment with ovalbumin. The horizontal axis is the number of amino acids, and the vertical axis is the size of fluctuation.
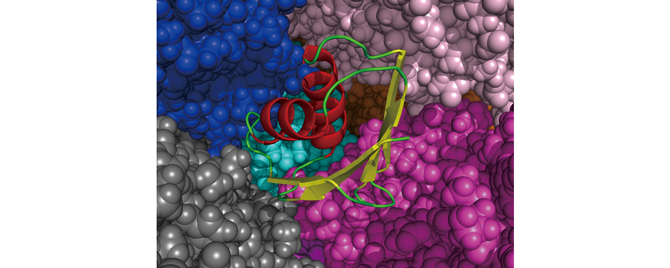
Fig 2 : TTHA surrounded by ovalbumin. The spiral in the center and arrows represent TTHA. The surrounding spherical shapes are ovalbumin atoms, where each different color represents one ovalbumin.
BioSupercomputing Newsletter Vol.6
- SPECIAL INTERVIEW
- Development of New Fluid-structure Interaction Analysis (ZZ-EFSI) Resulting in Rapid Achievement of High Operation Performance
Research Associate Professor, School of Engineering, The University of Tokyo Kazuyasu Sugiyama - Interview with High-performance Computing Team Members: Continued Efforts in Tuning to Harness the Potentials and the High Capability of the K Computer
Group Director of Computational Molecular Design Group,
Quantitative Biology Center, RIKEN
Makoto Taiji
Senior Researcher of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Yousuke Ohno
Senior Researcher of High Performance Computing Development Team,
High Performance Computing Development Group,
RIKEN HPCI Program for Computational Life Sciences
Hiroshi Koyama
Researcher of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Gen Masumoto
Research Associate of High-performance Computing Team,
Integrated Simulation of Living Matter Group,
Computational Science Research Program, RIKEN
Aki Hasegawa
- Report on Research
- Functional Analysis of Multidrug Efflux Transporter AcrB by All-Atom Molecular Dynamics Simulation
Graduate School of Nanobioscience,
Yokohama City University
Tsutomu Yamane,
Mitsunori Ikeguchi
(Molecular Scale WG) - Multi-scale Modeling of the Human Cardiovascular System
Computational Science Research Program,
RIKEN Liang Fuyou (Organ and Body Scale WG) - Toward a spiking neuron-level model of the early saccade visuomotor system
Kyoto University Jan Moren
Nara Institute of Science and Technology Tomohiro Shibata
Okinawa Institute of Science and Technology Kenji Doya
(Brain and Neural Systems WG) - Developing the MD Core Program for Large Scale Parallel Computing
Computational Science Research Program,
RIKEN Yousuke Ohno (High-performance Computing Team)
- SPECIAL INTERVIEW
- Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Program Director of RIKEN HPCI Program for Computational Life Sciences
Toshio Yanagida
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences
Akinori Kidera
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences
Yukihiro Eguchi
- Report on Research
- Free Energy Profile Calculations for Changes in Nucleosome Positioning with All-Atom Model Simulations
Quantum Beam Science Directorate, Japan Atomic Energy Agency
Hidetoshi Kono, Hisashi Ishida, Yoshiteru Yonetani, Kimiyoshi Ikebe (Field 1- Program 1) - Estimation of Skeletal Muscle Activity and Neural Model of Spinal Cord Reflex
Information Science and Technology, The University of Tokyo
Yoshihiko Nakamura (Field1 - Program 3)
- Report
- ISLiM Interim Accomplishment Meeting in 2011
Computational Science Research Program, RIKEN Eietsu Tamura - Computational Life Sciences Classes Held in High Schools
HPCI Program for Computational Life Sciences, RIKEN
Chisa Kamada, Yasuhiro Fujihara, Yukihiro Eguchi