

Prospects of Prognostic Prediction Based on Genome-wide Association
Study and Genetic/Non-genetic Factors

Riken Center for Genomic Medicine
(Data Analysis Fusion WG)
Naoyuki KAMATANI
Studies on the relationship between personal difference in genome sequences and traits have been rapidly promoted since the elucidation of the human genome in 2003. Traits refer to attributes that vary from person to person, for example, "being disease or non disease" or "responsiveness to a certain drug." In respect to Mendelian disorders, a method called "linkage analysis" has been established that can almost certainly identify causative genes if sufficient genealogic information is provided. Linkage analysis was first proposed by Fisher in 1922 with the use of the "maximum likelihood method," a mathematical approach developed by Fisher himself. Along with the development of numerous markers for the human genome and the improvement of computer performance, linkage analysis was quickly applied to the elucidation of genetic diseases.
Next, researchers became interested in multi-factorial traits. Multi-factorial traits do not have the Mendelian genetic form, but rather a complex genetic form and are presumed to be influenced by multiple genes and the environment. Traits can be classified into quantitative traits and qualitative traits, many of which have two phenotypes. The types of influences on qualitative and quantitative traits induced by multiple genes and the environment were formulated by Fisher in 1918 as an additive polygene model. Based on this model, linkage analysis using data on numerous (500,000-1,000,000) markers for the human genome is currently under way. This approach is known as the "Genome-wide Association Study (GWAS)" and it is a prominent method to understand the genetic factors of multi-factorial traits. GWAS was successfully adopted for the first time anywhere in the world by the Riken Center for Genomic Medicine (then called the SNP Research Center) in 2002.
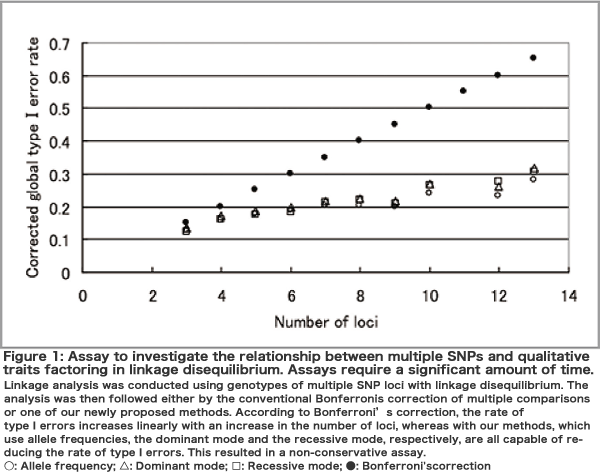
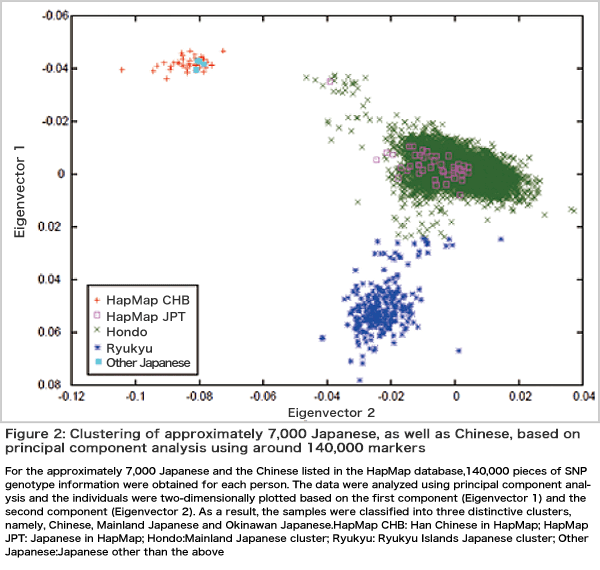
The most important task of GWAS is data cleaning. Since several hundred thousand pieces of information for each individual are obtained from hundreds or thousands of people, it takes a lot of work to clean the data involved. The second most important task is to perform an assay, i.e., to investigate whether there is a relationship between traits and genomic diversification. Here, the problem relating to multiple comparisons arises because the assay has to be conducted several hundred thousand times. The normal statistical significance of P < 0.05 is insufficient and the P value must be at the level of 10-7-10-8. We have developed and proposed an algorithm for performing linkage analysis with the use of numerous markers while taking into account linkage disequilibrium (Figure 1). Furthermore, an analysis of population structuring is also important, because it may lead to false positives. Based on a principal component analysis, we reported that Japanese are classified into two distinct clusters (mainland and Ryukyu clusters) and that people in the mainland cluster also have significant genetic differences depending on the areas where they live (Figure 2). The third most important task is the estimation of various parameters and the interpretation of the results. Finally, we develop algorithms that use data from various analyses to predict disease susceptibility and drug responsiveness and then evaluate these algorithms.
Each of the above steps is important, but many of them require a great deal of time for calculation. Moreover, longer calculation times are required as the number of samples or control markers increases. Normally, calculations are performed on the assumption that the influence of each gene or each environmental factor is independent, but the calculation time becomes even longer when interaction is factored in. Recently, the amount of genomic data that can be obtained from one person is increasing dramatically with the introduction of ultrafast sequencers. In short, although we have the data, we are unable to accomplish the task because the calculations take too long. It is certain that the genetic causes of diseases, which are presently unidentified, will be identified when petaflop computers become available.
Despite the huge amount of data obtained as described above, prediction accuracy is usually not as high as one might expect due to the unstable probability. Probability in the laws of genetic inheritance is quite stable, however, ensuring the accuracy of prognostic prediction based on genomic data.
BioSupercomputing Newsletter Vol.1
- SPECIAL INTERVIEW
- Innovative Approach for Understanding Phenomena of Life Exploring New Possibilities with Bio-supercomputing
Computational Science Research Program Deputy Program Director Ryutaro HIMENO
- A Message from the Team Leader
- Simulations to Understand the Functions of the Biopolymers that Play Fundamental Roles in Life
Molecular Scale Team Team Leader Akinori KIDERA - Develop a 3-D Model of the Entire Human Body and Understand In Vivo Phenomena to Utilize for Medical Purposes
Organ and Body Scale Team Team Leader Shu TAKAGI - The Fourth Methodology (Data Analysis Fusion): Transforming Biology into a Predictable Science
Data Analysis Fusion Team Team Leader Satoru MIYANO
- Report on Research
- Prediction of Transmembrane Dimer Structure of Amyloid Precursor Protein using Replica-Exchange Molecular Dynamics Simulations
Molecular Scale Team Naoyuki MIYASHITA / RIKEN Advanced Science Institute (Molecular Scale WG) Yuji SUGITA - Simulation for Charged Particle Therapy
Organ and Body Scale Team Kenichi L. ISHIKAWA - Prospects of Prognostic Prediction Based on Genome-wide Association Study and Genetic/Non-genetic Factors
Riken Center for Genomic Medicine (Data Analysis Fusion WG) Naoyuki KAMATANI - Key Technology Supporting Petascale Computing
High-performance Computing Team Kenji ONO / Satoshi ITO / Daisuke WATANABE