

Prediction of Transmembrane Dimer Structure of Amyloid Precursor Protein using
Replica-Exchange Molecular Dynamics Simulations


Molecular Scale Team
Naoyuki MIYASHITA (left)
RIKEN Advanced Science Institute
(Molecular Scale WG)
Yuji SUGITA (right)
Alzheimer's disease is a neurodegenerative disorder and its main symptom is to disturb cognitive functions. In the progressive process of this disease, the death of nerve cells in the brain (neurons) occurs. The most widely accepted hypothesis for the cause is the amyloid hypothesis, in which Alzheimer's disease results from the aggregation and accumulation of amyloid β(Aβ) peptide in the brain. Therefore, it is vitally important to acquire better knowledge of the formation of Aβ peptides in understanding Alzheimer's disease.
Amyloid precursor protein (APP) is composed of approximately 700 amino-acid residues in the membrane of neurons. Aβ denotes a series of approximately 40 residues in APP and is derived from the sequential cleavage at both ends of these consecutive residues (upper and lower) byβ- andγ-secretases, respectively. Aβ1-40 consisting of 40 aminoacid residues is a primary isoform, whereas a varyingγ-cleavage site can produce Aβ1-42, which is a major component of senile plaques. How can these different lengths of Aβ peptides be produced from APP? The transmembrane structure of APP has not been revealed by X-ray crystallography, presumably due to the large fluctuations of APP. We attempt to predict atomic structure of APP in the membrane, using molecular dynamics simulations and compare the results with recent biochemical experiments.
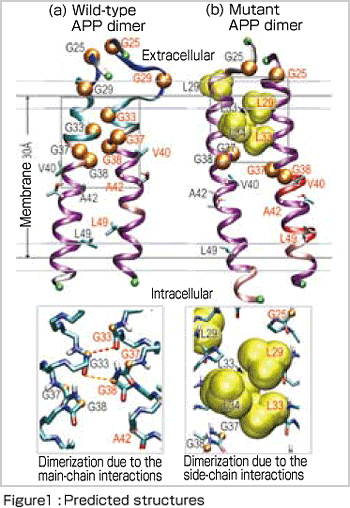
In this study, we focus on Aβ23-55, in the transmembrane (TM) and juxtamembrane regions , because contains key residues in APP. Aβ23-55 includes five consecutive glycine (Gly) residues and is considered to form a pair (dimer) in the membrane. In fact, recent experiments have demonstrated dimerization of wild-type APP (having a normal amino-acid sequence) in the intramembrane. Interestingly, it has been reported that when two Gly residues (Gly29 and Gly33) out of five are replaced with hydrophobic* leucine (Leu) residues, this mutant also forms a dimer as in wild-type APP but cannot be cleaved byγ-secretase (which resuts in no production of Aβpeptides)[1]. How can the mutations of only two residues in the amino -acid sequence affect the structure and function of APP ?
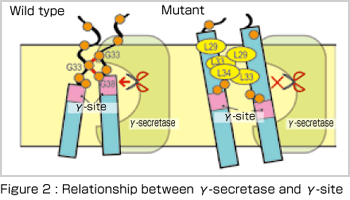
To answer this question, it is usually effective to perform molecular dynamics simulations that can deal with molecular interactions in protein. If atomic structure of the relevant protein is not determined by X-ray crystallography, a thermodynamically stable conformation selected from various possible conformations by computer simulations can be the basis for discussion. Generalized-ensemble algorithms, including the replica-exchange molecular dynamics method[2] used in this study, are especially suitable for this purpose and many new methods have been developed by researchers in Japan, such as Professor Yuko Okamoto in Nagoya University. Our results[3], as shown in Figure 1, indicate the differences in dimer formation between wild-type and mutant APPs. In the mutant APP, the increase in the number of hydrophobic amino-acid residues causes APP to tilt toward the membrane and thus the site for cleavage in APP no longer matches the active site of γ -secretase [Figure 2]. The calculation results are compatible with the recent experimental data and moreover the molecular mechanism suggested by us has attracted the interest of many experimental researchers.
In this study, because of the limitation of computational resources available at present, we do not explicitly include solvent and phospholipid molecules, but rather use a model in which the influence of solvent and membrane are included implicitly, considering the effect of the excluded volume. In the next-generation supercomputer, we can include solvent and phospholipid molecules in the simulations of membrane proteins and expect to obtain more reliable simulation results. The replica-exchange molecular dynamics simulation is a promising methodology in the nextgeneration supercomputer, because it can effectively simulate substrate binding or conformational changes in large proteins as the number of CPUs available increases. In addition to Alzheimer's disease, phenomena involved in many other diseases are also associated with the behavior of membrane proteins. We hope that our studies will lead to a better understanding of the biological activities associated with various diseases and can be utilized for their treatments.
* Note : Twenty kinds of amino acids, which constitute proteins, share a common structure called the main chain, in which a hydrogen atom, an amide group (NH2) and a carbonyl group (CO) are bound to a carbon atom (Cα) in the center. Each amino acid is characterized by its side chain that is also bound to the Cα atom. Twenty kinds of side chains can be categorized into two groups: hydrophilic, with a property of attracting water and hydrophobic, with a property of repelling water.
References
[1] P. Kienen-Campard et al., J. Biol. Chem. 283, 7733-7744, (2008)
[2] Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 314, 141-151, (1999)
[3] N. Miyashita, J.E. Straub, D. Thirumalai and Y. Sugita, J. Am. Chem. Soc.,
131, 3438-3439, (2009)
BioSupercomputing Newsletter Vol.1
- SPECIAL INTERVIEW
- Innovative Approach for Understanding Phenomena of Life Exploring New Possibilities with Bio-supercomputing
Computational Science Research Program Deputy Program Director Ryutaro HIMENO
- A Message from the Team Leader
- Simulations to Understand the Functions of the Biopolymers that Play Fundamental Roles in Life
Molecular Scale Team Team Leader Akinori KIDERA - Develop a 3-D Model of the Entire Human Body and Understand In Vivo Phenomena to Utilize for Medical Purposes
Organ and Body Scale Team Team Leader Shu TAKAGI - The Fourth Methodology (Data Analysis Fusion): Transforming Biology into a Predictable Science
Data Analysis Fusion Team Team Leader Satoru MIYANO
- Report on Research
- Prediction of Transmembrane Dimer Structure of Amyloid Precursor Protein using Replica-Exchange Molecular Dynamics Simulations
Molecular Scale Team Naoyuki MIYASHITA / RIKEN Advanced Science Institute (Molecular Scale WG) Yuji SUGITA - Simulation for Charged Particle Therapy
Organ and Body Scale Team Kenichi L. ISHIKAWA - Prospects of Prognostic Prediction Based on Genome-wide Association Study and Genetic/Non-genetic Factors
Riken Center for Genomic Medicine (Data Analysis Fusion WG) Naoyuki KAMATANI - Key Technology Supporting Petascale Computing
High-performance Computing Team Kenji ONO / Satoshi ITO / Daisuke WATANABE