

Molecular Scale Team
Simulations to Understand the Functions of the Biopolymers that
Play Fundamental Roles in Life

Molecular Scale Team
Team Leader
Akinori KIDERA
Genes can be found on the bottom level of biological hierarchy, while individual living organisms are at the top. The Molecular Scale Team has been researching the world of molecules, which come closest to the bottom level. Genes can be compared to hard disks in which an amount of information is accumulated. In order for genes to actually perform the biological activities, they need to be transformed into something functional; more specifically, proteins. Molecules include DNA or genes, in other words, as well as the RNA that is generated in the process of transcription and a variety of relevant components, but proteins are the main players among molecules. Proteins come together to perform a variety of functions. Cells can be found at the top of the bottom level, forming tissues and organs and ultimately forming individual living organisms. Our main challenge is to clarify what kind of roles proteins play in individual living organisms. Our aim is to understand the world of the molecules that form the fundamental basis for the activities of life at the most basic level as is possible by virtually activating the molecules in the supercomputer with fully utilizing the molecular simulation methods.
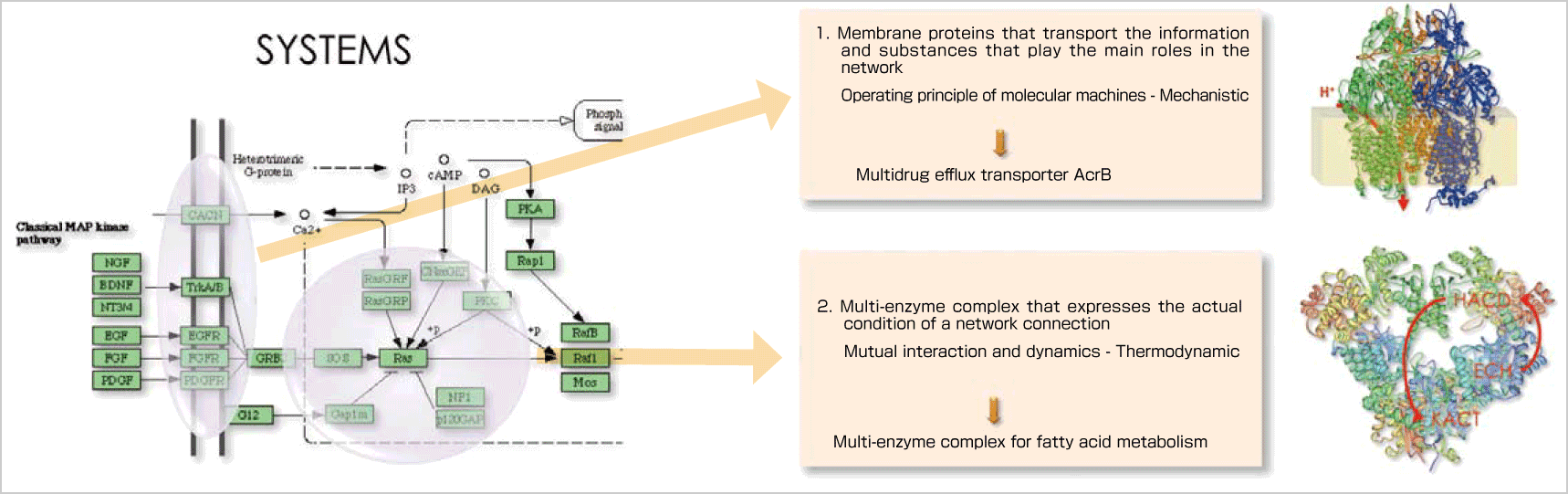
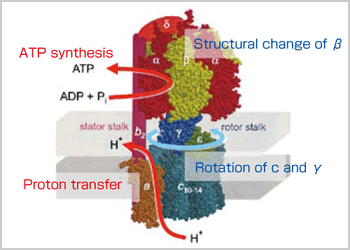
As an example of molecular simulation methods, I'd like to explain ATP synthase. ATP (adenosine triphosphate) is the most important energy resource for living organisms. Energy extracted from organic matter is stored in the form of the energy compound ATP and energy generated at the time of ATP breakdown, which occurs on an as-needed basis, is utilized for a variety of the biological activities. ATP synthase is involved in the process of synthesizing ATP. A synthase is a form of molecular machine made of proteins and features sophisticated functions. The synthase remains stuck in the cell membrane. The transfer of hydrogen ions from the outside toward the inside (proton transfer) generates a rotating force on the synthase, which is transformed to the energy needed for protein conformational change. This energy is used for synthesizing ATP. These functions have been confirmed through experiments and we intend to explain the principles of the functions through molecular simulations. There are two main methods for understanding these functions. One is the quantum chemistry calculation (QM), which can handle first principle calculations of chemical reactions. The other is the classical level molecular dynamics calculation (MM), which can calculate the movements. In respect to ATP synthase, proton transfer, for example, is a reaction that should be handled by quantum chemistry. The rotations of macromolecules are dynamic movements and can be understood through molecular dynamics calculations. The conformational changes can also be understood through molecular dynamics, and the subsequent chemical reaction between phosphoric acid and ADP should be through quantum chemical calculations. By utilizing these two methods, the principles of the ATP synthase functions can be understood. Computer experiments are carried out by constructing protein complexes, and activating them to initiate the reactions , such as ATP synthesis. Through these procedures, we can clarify reactions under various conditions, including the responses of protein complexes to certain external influences, resulting in new discoveries and theories that cannot be achieved through experimentation.
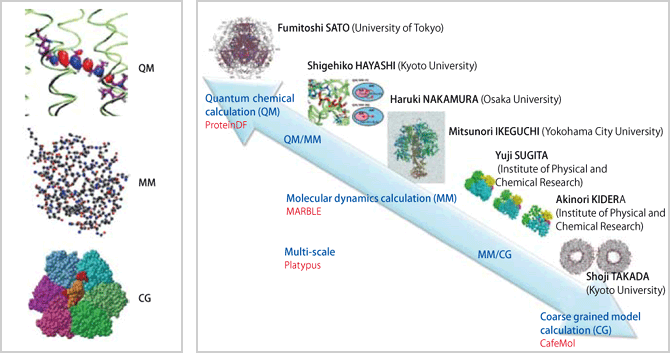
The problem here is computer resources. In order to carry out the aforementioned simulations on a computer, a huge amount of calculations are required. Even if we use a supercomputer, it would be impossible to calculate everything starting from quantum chemical calculation or even from molecular dynamics simulation. To overcome this difficulty, we have been developing another method – coarse grained (CG) model calculation – in order to see the overall picture of the functional expression of proteins. For instance, the properties of each of the amino acids that comprise proteins are modeled, replacing each of the amino acids with a sphere specified with certain interaction parameters. We are trying to express biomolecular systems in three levels, specifically QM, MM and CG. The CG model calculation is a new method in the field of molecular simulation. The major issues for our team include the establishment of coarse graining methodologies, the development of new software and the development of methodologies for multi-scale simulations that are carried out by combining the three methods of QM, MM and CG (coupled simulation connecting different levels of biological hierarchy). Our main and ultimate goal is to lead our molecular level efforts into an understanding of cells, one layer above the molecular level. In other words, we are trying to explain phenomena in cells from the molecular level.
BioSupercomputing Newsletter Vol.1
- SPECIAL INTERVIEW
- Innovative Approach for Understanding Phenomena of Life Exploring New Possibilities with Bio-supercomputing
Computational Science Research Program Deputy Program Director Ryutaro HIMENO
- A Message from the Team Leader
- Simulations to Understand the Functions of the Biopolymers that Play Fundamental Roles in Life
Molecular Scale Team Team Leader Akinori KIDERA - Develop a 3-D Model of the Entire Human Body and Understand In Vivo Phenomena to Utilize for Medical Purposes
Organ and Body Scale Team Team Leader Shu TAKAGI - The Fourth Methodology (Data Analysis Fusion): Transforming Biology into a Predictable Science
Data Analysis Fusion Team Team Leader Satoru MIYANO
- Report on Research
- Prediction of Transmembrane Dimer Structure of Amyloid Precursor Protein using Replica-Exchange Molecular Dynamics Simulations
Molecular Scale Team Naoyuki MIYASHITA / RIKEN Advanced Science Institute (Molecular Scale WG) Yuji SUGITA - Simulation for Charged Particle Therapy
Organ and Body Scale Team Kenichi L. ISHIKAWA - Prospects of Prognostic Prediction Based on Genome-wide Association Study and Genetic/Non-genetic Factors
Riken Center for Genomic Medicine (Data Analysis Fusion WG) Naoyuki KAMATANI - Key Technology Supporting Petascale Computing
High-performance Computing Team Kenji ONO / Satoshi ITO / Daisuke WATANABE