
 Strategic Programs for Innovative Research
Strategic Programs for Innovative Research
Field 1 Supercomputational Life Science
Theme 4 Large-scale analysis of life data
Leading-edge large-scale sequence data analysis with K computer in order
to promote the understanding of life programs and their diversity

Professor, Human Genome Center, The Institute of Medical Science
The University of Tokyo
Satoru Miyano
(Theme4 GL)
●Elucidation of life systems through large-scale data analysis
In Theme 4 “Large-scale analysis of life data”, after establishing an infrastructure for leading-edge large-scale sequence data analysis optimized for High Performance Computing Infrastructure (HPCI) centering on the K computer, we are proceeding with research, in which we consider life programs found in cancers and cell differentiation processes as a system and understand their complexity and diversity based on the genomes. We also conduct biomolecule network analysis research centered on the data based on genomes. Through this research, we aim to contribute to practical applications such as better predictions of drug efficacy and adverse reactions, projections of causes of toxicities, personalized medicine, and prediction of survival.
“Life data” in the title of course includes the genome. It also covers a massive amount of data based on the genomes such as the epigenome (the entire set of DNA modifications) and transcriptome (all transcription products). They are key data for understanding life programs.
As an example to understand their importance, let’s consider why we get cancer. Cancer is a kind of disorder in life systems induced by the complex combination of genetic factors inherited from parents, DNA modification (epigenome) by environmental factors and genetic mutation accumulated in tumor cells. Systems disorder may result in malfunction of a selfdestruction system for the prevention of abnormality (apoptosis) or selfordering for growth due to malfunction of external order for arresting growth. In addition, cancer cells join hands with normal cells such as vascular endothelial cells, immunocytes and inflammatory cells to acquire drug resistance, or spread anywhere through infiltration and metastases. Due to this, cancer is a complex and abnormal cell group that evolves spatiotemporally. It is the genome that regulates the malignancy grading of cancer, responsiveness to treatment and likeliness to have adverse reactions. In order to elucidate systems disorder, we have to examine the genome, epigenome, trascriptome as well as proteome (the entire set of proteins), metabolome (all metabolic products), and interactome (the entire set of interactions). However, we cannot understand a treatment approach or state of disease with these alone. For elucidating the diversity, complexity and dynamism of cancer, it is crucial to understand the system by large-scale data analysis and mathematical modeling taking advantage of mathematics and supercomputers.
●Breakthrough for understanding life systems through mathematical analysis by use of the K computer
In 2003, the 13-year “Human Genome Project”, which cost 100 billion yen, finally succeeded in decoding the human genome. This year, the National Institutes of Health (NIH) provided a roadmap after decoding the human genome, which says “Biology is changing fast into a Science of Information Management.” In 2003, it surprised many Japanese researchers, who thought it was an exaggerated claim. Now, 10 years since then, everyone keenly feels that the claim is coming to be realized. From 2003, the International HapMap Project to elucidate individual differences on a DNA level was put into practice mainly in the USA, and a basis for efficiently finding genes involved in human diseases and drug responsiveness was established. The International Cancer Genome Consortium started to prepare a genomic aberration catalogue of major cancers from 2008, and has analyzed whole genome information on 50 thousand human genomes, consisting of cancer samples and normal cells of 25 thousand humans. Meanwhile, Barack Obama who assumed the presidency in 2008 introduced “The Genomics and Personalized Medicine Act” when he was a senator. In 2009, “the Genetic Information Nondiscrimination Act” was enacted. Consequently, the USA steadily implemented medical and healthcare strategies based on genomic information. NIH started to allocate its budgets to the $1,000 Genome (a project to drop the cost of full genome sequencing per human to roughly $1,000) from as early as 2004. Thanks to this, development and practical application of sequencing technology was promoted, and now Biology is becoming a science to analyze and interpret massive amounts of genome data.
These days, sequencing technology is advancing rapidly. The nextgeneration sequencer using conventional optical sequencing technology is an expensive instrument, and also has some problems such as need of extravagant fluorescent reagents and limited lead length. However, today, cutting-edge sequencers such as a semiconductor sequencer using lowcost silicon chips have been developed, and an instrument which can thousand within several hours” will be put to practical use in 2013. It will give steady service a couple of years later. Moreover, sequencers providing personal genome information “at $100 within an hour” are said to be “around the corner”. With the spread of super-low price, high-speed, high-precision sequencers, personalized medicine, in which disease diagnosis and selection of therapeutic strategy and type and dosage of the drug are decided based on clinical sequence to check your own DNA, will rapidly become popular. I believe the day is not far off when everyone has his or her own DNA sequence. When the personal genome era begins in earnest, an enormous amount of genome data will be produced, and new breakthroughs will be applied to the understanding of life systems.
The first results have already been produced. It is the “discovery of causative genes for chronic myelodysplastic syndrome (MDS).” Many cases of MDS transform to acute myelocytic leukemia since bone marrow cannot produce normal blood. Although its cause had been unknown, the team of Seishi Ogawa (Cancer Board, University of Tokyo Hospital) and his colleagues fully employed the supercomputer system at Human Genome Center (The Institute of Medical Science, The University of Tokyo) and discovered from the thorough analysis of patient samples’ exomes (remnant after an RNA splicing reaction) using next-generation sequencers that the cause was mutations in 4 genes in the RNA splicing pathway. This is said to be a historic result in cancer research, since it is not only the first discovery of the causative genes for this disease, but has also shown that abnormal RNA splicing plays a role in the development of cancer for the first time in the world. This can moreover be said to be the result of a successful combination of sequencing technology, supercomputer and a statistical mathematics analysis team. Also in the “Large-scale analysis of life data” of Strategic Programs for Innovative Research Field 1, in which we are now engaged, we would also like to achieve such breakthroughs.
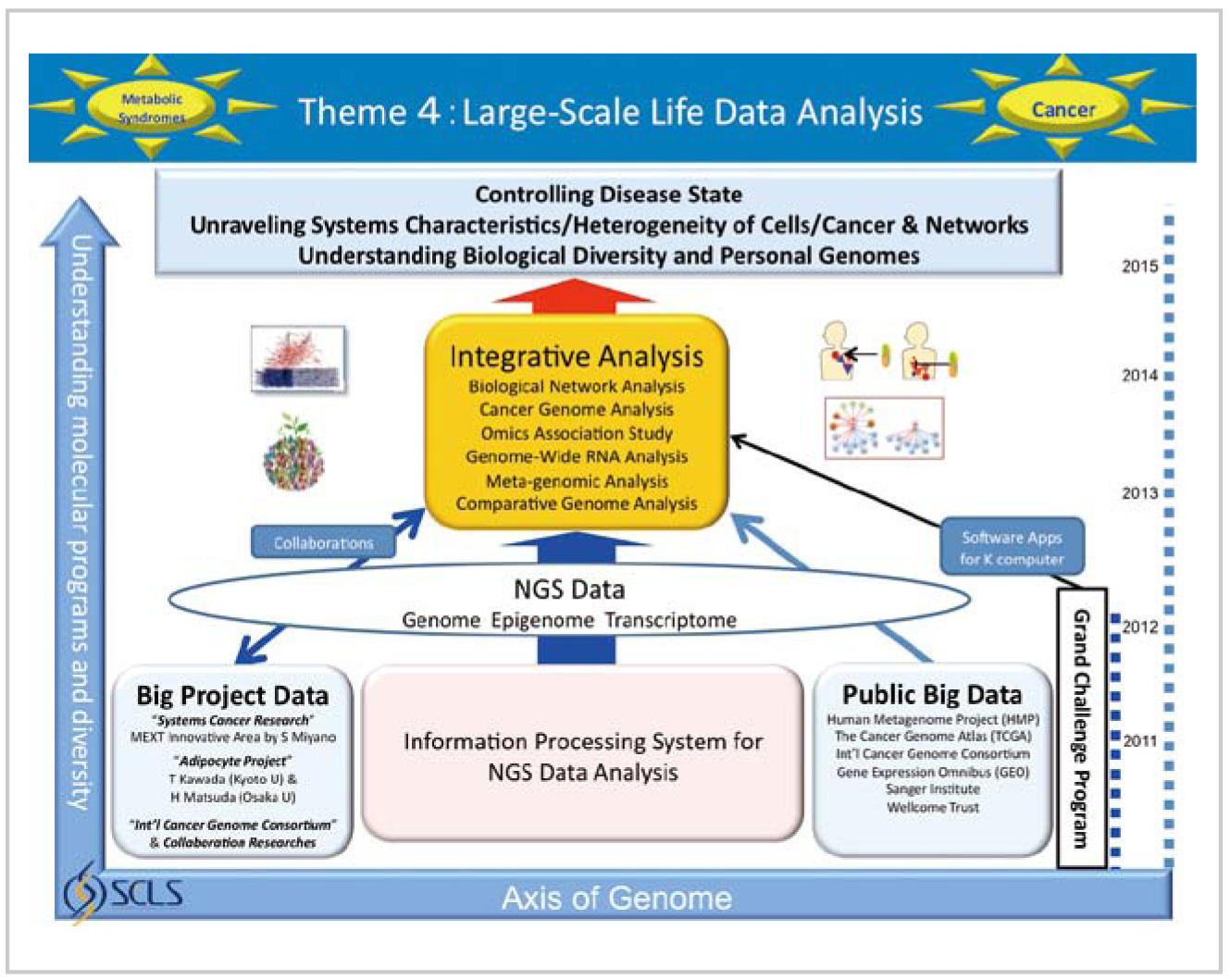
Research advancement plan of Theme 4
●Content of the research Theme 4 address
The major objective of Theme 4, “Large-scale analysis of life data,” is the understanding of the life program and its diversity. The first thing required to achieve this goal is the high-speed processing of massive amounts of next-generation sequencer data. For this purpose, we made great efforts to establish the basis for highspeed analysis of massive amounts of next-generation sequencer data optimized for “the K computer” from 2011 to 2012. In this analysis system, we are trying to realize high-speed analysis of massive genome sequence information by achieving the performance of 10 million reads per hour and performing the world`s deepest high-speed sequence searching which can detect more challenging smaller homology. In this way, we are aiming to create a system to ensure a 10-to-1 advantage over the rest of the world.
The basic data for the analysis includes not only public data but also the Cancer TCGA Project and the Metagenome Project data, and we collaborate with the International Cancer Genome Consortium which researchers of Theme 4 participate in, the Innovative Areas “Systems Cancer Research” and “Adipocyte Project.” Validation by experiment is also carried out in those projects.
The specif ic content of the research is roughly grouped into “Understanding of the lineage of cancer and its diversity,” “Deepening of personal genome understanding,” “Personal medical intervention prediction,” “Cell differentiation network,” “Drug response network” and “Personal caner network.” Although the research will be extensive, it has 3 goals. One is “Deepening the understanding of the biological diversity and personal genomes.” We especially focus on the metagenome and the diversity of cancer. In addition, by advancing the understanding of personal genomes, we believe we can deepen understanding of the biological diversity. The second is “Unraveling systems characteristics/heterogeneity of cells/cancer and networks.” The third is “Control of disease state” by leading those findings to prediction of medical intervention. We are going to advance our own research to achieve those three goals.
Simulation studies are being vigorously performed in Life Science. However, the life system has not yet been sufficiently clarified to simulate even E . c oli fully. Therefore, although understanding of the human life system is advancing, it is probably still very difficult nowadays to draw new biological findings or develop a drug effective for everyone by constructing a mathematical model for simulation. In Theme 4, we start from data for data-driven science. It is our approach to develop methods to seek a clue to the cause for disease of each individual by analyzing data in an integrated way, and identifying something visible from that.

MicroRNA/mRNA gene network of lung adenocarcinoma. Overall view of the causal relations among genes (by courtesy of Professor Takashi Takahashi (School of Medicine, Nagoya University)).
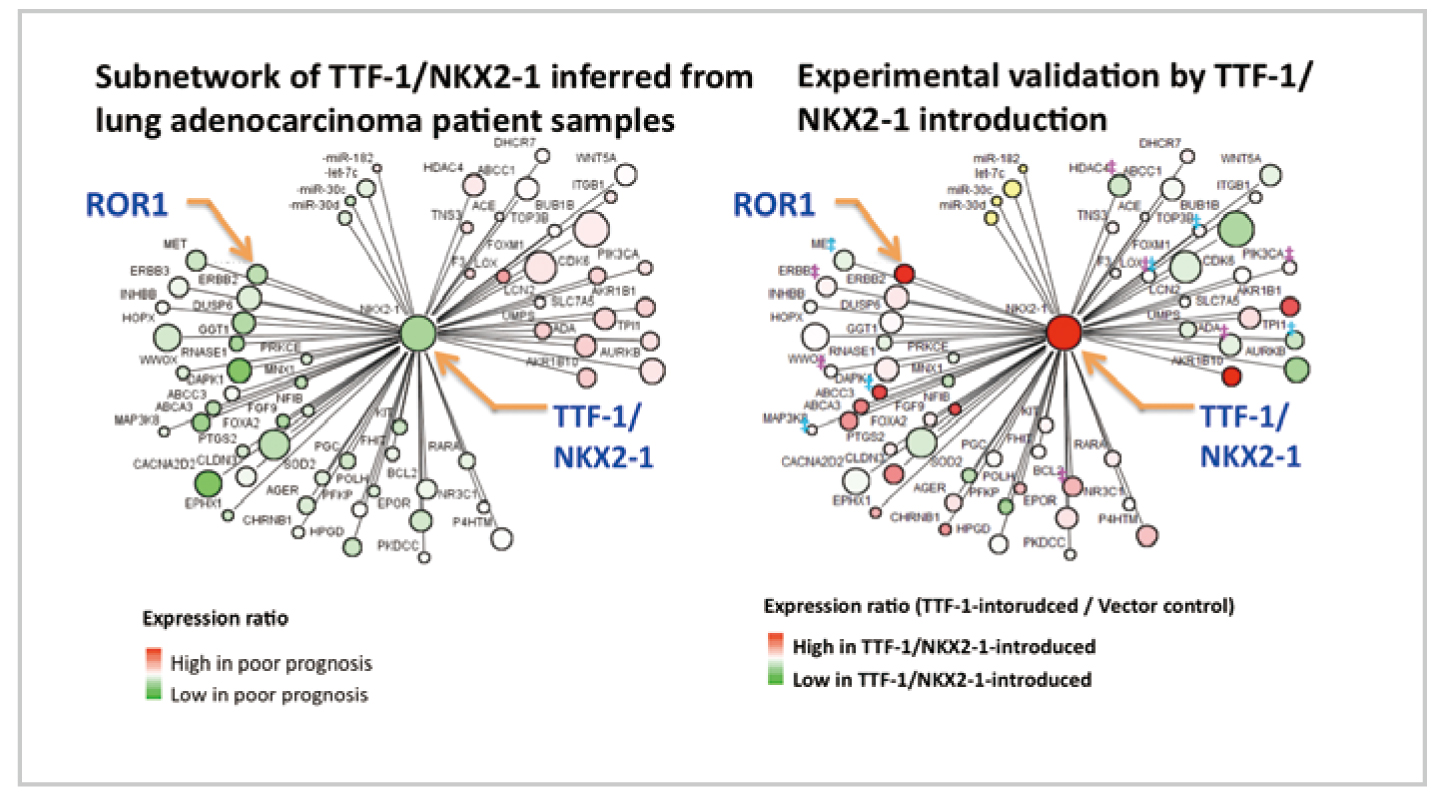
Introduction of the gene TTF-1/NKX2-1 changed the gene expression of surrounding network that shows a significant relationship to survival. It is seen that the switch to determine whether or not lung adenocarcinoma survives has been altered (by courtesy of Professor Takashi Takahashi (School of Medicine, Nagoya University)).
BioSupercomputing Newsletter Vol.8
- SPECIAL INTERVIEW
- Grand Challenge opens the way to the future of life science through innovative approach
Program Director, Computational Science Research Program, RIKEN
Koji Kaya - A Landmark Project that Brought on an Innovation in the Field of Life Scienc
Deputy-Program Director, Computational Science Research Program, RIKEN
Ryutaro Himeno
- Report on Research
- Multi-scale, multi-physics heart simulator UT-Hear
Graduate School of Frontier Sciences, the University of Tokyo
Toshiaki Hisada, Seiryo Sugiura, Takumi Washio,
Jun-ichi Okada, Akihito Takahashi
(Organ and Body Scale WG) - Simulation Model for Insulin Granule Kinetics in Pancreatic Beta Cells
Graduate School of System Informatics, Kobe University
Hisashi Tamaki(Cell Scale WG) - The road to brain-scale simulations on K
Brain Science Institute, RIKEN, Institute of
Neuroscience and Medicine (INM-6),
Juelich Research Center
Medical Faculty, RWTH Aachen University Markus Diesmann
(Brain and Neural Systems WG) - MD Core Program Development for Large-scale Parallelization
Computational Science Research Program, RIKEN
Yousuke Ohno(High-performance Computing Team)
- SPECIAL INTERVIEW
- Aiming to realize hierarchical integrated simulation in the circulatory organ system
and the musculoskeletal / cerebral nervous systems
Professor, Department of Mechanical Engineering and Department of Bioengineering The University of Tokyo
Shu Takagi(Theme3 GL) - Leading-edge large-scale sequence data analysis with K computer in order to promote the understanding of life programs and their diversity
Professor, Human Genome Center, The Institute of Medical Science, The University of Tokyo
Satoru Miyano(Theme4 GL)
- Report
- 4th Biosupercomputing Symposium Report
Computational Science Research Program, RIKEN
Eietsu Tamura - “K Computer” Compatible Computer: Installation of SCLS Computer System
HPCI Program for Computational Life Sciences, RIKEN
Yoshiyuki Kido