

MD Core Program Development
for Large-scale Parallelization

Computational Science Research Program, RIKEN
Yousuke Ohno
(High-performance Computing Team)
Our team has been developing a MD core program for large-scale parallelization in order to provide a high speed core program library for the "K computer", and to attain and collect application speedup technology knowhow on the "K computer".
Molecular dynamics (MD) simulation is a method for estimating the movement of molecules and changes in structure through calculation. In life sciences, biomolecules such as protein which are the foundation of life phenomena are targets of calculation. In the simulations, all movements of atoms are calculated by calculation of the all forces between atoms and numerically integration of the equation of motion. The amount of calculation for the numerical integration of the equations of motion and forces of covalent bond approximated by classical is proportional to the number of atoms. On the other hand, Van der Waals forces (intermolecular forces) and Coulomb forces incur a calculation load that is proportional to the number of pairs of atoms, which means the square of the number of atoms. Van der Waals forces are very small after a distance of 1.4nm, therefore pairs of atoms that are distant are ignored which is called a cutoff method. However, Coulomb force is inversely proportional to the square of the distance, and cannot be blindly ignored. Therefore the PME (Particle Mesh Ewald) method, which reduces the calculation load of long distance Coulomb force as low as O (N log N ) by using FFT, is a popular way to calculate Coulomb force. Yet, FFT requires a high amount of communication when parallelized, which leads to lower performance in large-scale parallelization. Therefore, methods like the FMM (Fast Multiple Method) which demands less communication compared to FFT are starting to be employed. In addition, the PME method is very strict under periodic boundary conditions, and intracellular targets which do not have periodic cycles may have problems in using a pseudo-period. In that case, a cutoff method that corrects electrical charge and dipole moments under neutral conditions may be applied [1, 2]. The cutoff method requires communication between adjacent nodes, which is suitable for large-scale parallelization.
In our research, optimizing a cutoff method on the "K computer" became the center of our study because some form of cutoff calculation is the major part of calculation time regardless of the methods used. We attained a calculation efficiency of 60% for the cutoff calculation portion on a single CPU using a cutoff method with the best conditions. In the all node evaluations of the "K computer" (Note 1) in October 2012, we attained 4 PFlops effective calculation speed and 38% efficiency on 5.4 hundred million atoms with a cutoff distance of 2.8nm and once per four-step potential calculation. When potential is calculated in each step, effective calculation speed was 4.4 PFlops and 43% efficiency for a calculation of a 79872 node with 5.2 hundred million atoms.
Fig. 1 shows the parallel performance. The green dotted line represents parallelism when the calculation size (number of atoms) per node is constant (weak scaling). The number of atoms per node is 52338, 6542, 818, 102 in descending order. Calculation time is fixed in every case and a high level of parallelism is attained. The red line represents parallelism when the entire calculation size is fixed (strong scaling) and the number of atoms is 418707, 3349656, 7536726, 26797248, 214377984 from left to right. Parallelism depends on the number of atoms per node, and performance improved proportionally to the number of nodes when there were 6000 and more atoms. But when the numbers of nodes were multiples of 8 or 64 and the numbers of atoms were 800 or 100 per node, parallel performance fell to 50% or 20%. Ten milliseconds for calculating one step on a node of 100 or so atoms seems to be the margin for practical use. Regarding the test evaluation of long distance Coulomb calculation using FMM, parallel performance did not weaken as much compared to cutoff calculations. Further, we confirmed that performance levels did not weaken much on a scale that is equivalent to a scope using all nodes of the "K computer".
The optimization on the "K computer" attained in our research has been applied in the development of applications and optimizing by other teams. Further, this MD core program will be released either as an example and a reference code for the optimization on the "K computer", or as an optimized MD code that can be reused.
Our research is joint research project with the High-performance Computing Team whose members are Hiroshi Koyama*, Gen Masumoto, Aki Hasegewa, Gentaro Morimoto, Noriaki Okimoto, and Hidenori Hirano.
*Hiroshi Koyama is now a researcher at National Institute for Materials Science (NIMS).
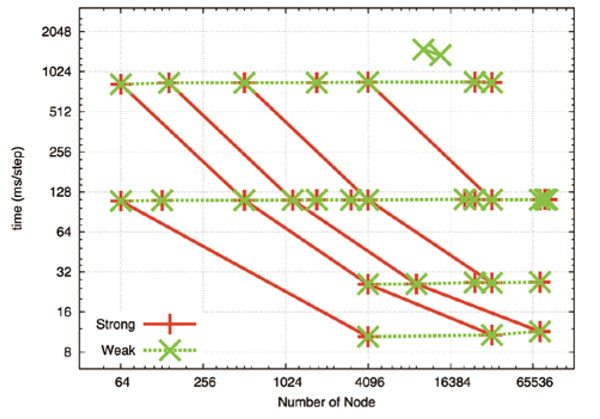
Fig. 1 : Parallel performance of the cutoff method. Cutoff distance of 2.8nm. Horizontal axis represents number of nodes, vertical axis represents calculation time per step (milliseconds). The green dotted line is a fixed number of atoms per node (weak scaling), the red line is when the number of all atoms is fixed (strong scaling).
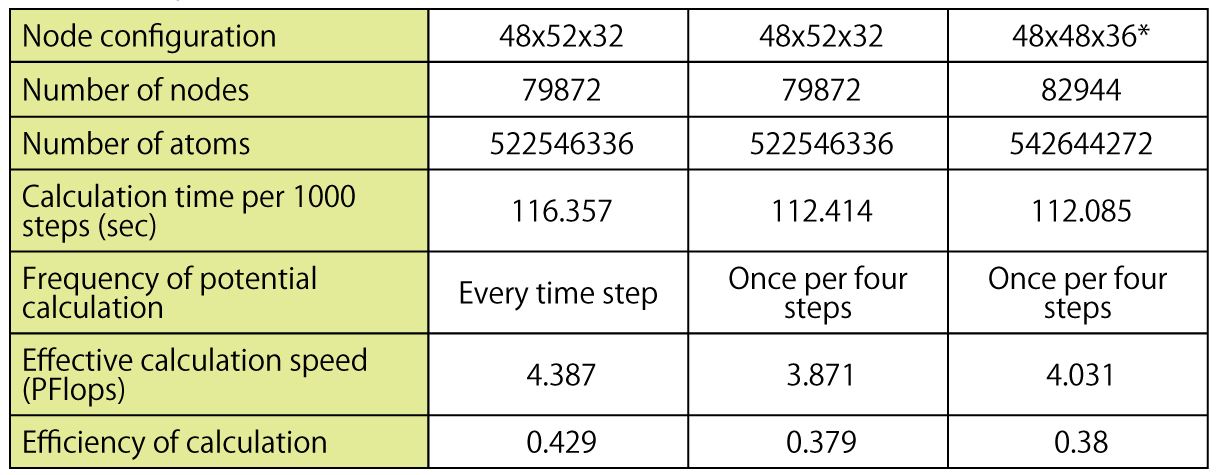
Table 1: Calculation performance of the cutoff method on a scale worth all nodes of the "K computer". Cutoff distance of 2.8nm. * Note: It differs from the actual network configuration of the "K computer" which is 48x54x32, but this does not greatly affect the results.
Note 1
Use of RIKEN's super computer "K computer" as a research program using the HPCI system (program number:hp120068)
[References]
[1] Wolf, D. and Keblinski, P. and Phillpot, SR and Eggebrecht, J., J. Chem. Phys. 110 8254 (1999)
[2] Fukuda, I. and Yonezawa, Y. and Nakamura, H., J. Chem. Phys. 134, 164107 (2011)
BioSupercomputing Newsletter Vol.8
- SPECIAL INTERVIEW
- Grand Challenge opens the way to the future of life science through innovative approach
Program Director, Computational Science Research Program, RIKEN
Koji Kaya - A Landmark Project that Brought on an Innovation in the Field of Life Scienc
Deputy-Program Director, Computational Science Research Program, RIKEN
Ryutaro Himeno
- Report on Research
- Multi-scale, multi-physics heart simulator UT-Hear
Graduate School of Frontier Sciences, the University of Tokyo
Toshiaki Hisada, Seiryo Sugiura, Takumi Washio,
Jun-ichi Okada, Akihito Takahashi
(Organ and Body Scale WG) - Simulation Model for Insulin Granule Kinetics in Pancreatic Beta Cells
Graduate School of System Informatics, Kobe University
Hisashi Tamaki(Cell Scale WG) - The road to brain-scale simulations on K
Brain Science Institute, RIKEN, Institute of
Neuroscience and Medicine (INM-6),
Juelich Research Center
Medical Faculty, RWTH Aachen University Markus Diesmann
(Brain and Neural Systems WG) - MD Core Program Development for Large-scale Parallelization
Computational Science Research Program, RIKEN
Yousuke Ohno(High-performance Computing Team)
- SPECIAL INTERVIEW
- Aiming to realize hierarchical integrated simulation in the circulatory organ system
and the musculoskeletal / cerebral nervous systems
Professor, Department of Mechanical Engineering and Department of Bioengineering The University of Tokyo
Shu Takagi(Theme3 GL) - Leading-edge large-scale sequence data analysis with K computer in order to promote the understanding of life programs and their diversity
Professor, Human Genome Center, The Institute of Medical Science, The University of Tokyo
Satoru Miyano(Theme4 GL)
- Report
- 4th Biosupercomputing Symposium Report
Computational Science Research Program, RIKEN
Eietsu Tamura - “K Computer” Compatible Computer: Installation of SCLS Computer System
HPCI Program for Computational Life Sciences, RIKEN
Yoshiyuki Kido