

An Ultra-fast Analysis System
for Next-Generation DNA Sequencer Data
HPCI Strategic Program Field 1 Supercomputational Life Science
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
(From the left) Yutaka Akiyama, Takashi Ishida, Masanori Kakuta, Shuji Suzuki (Field 1- Program 4)




The Next-Generation DNA Sequencer allows DNA sequencing of about 1 trillion bases with a single run by a dramatic technological innovation. Although the "number of bases sequenced per dollar" doubled with a marvelous extension rate in about 19 months with what we call a firstgeneration sequencer, it doubled in only about 5 months after the appearance of Next-Generation Sequencers which are coming into wide use everywhere. The largest bottleneck now is the calculation of semantic interpretation for read sequences. It is common knowledge that supercomputers are required for integrated advanced calculation such as gene network prediction. But now, large-scale calculations are also required for elementary processes for DNA fragments from sequencers.
A high-speed algorithm which is suitable to explore an exact match with a known sequence or highly similar character string can be used for the study of subtle variations of human genes, and large-scale personal computer clusters have managed to address it. However, in the study of a comprehensive analysis of enteric microbe's genome fragments of humans or the study of significant sequence mutations, fragment mapping procedure (finding correspondence with a portion of known genome sequence) is not possible without detection of sequence similarity among distant or largely mutated sequences. For these studies, incomparably large-scale calculation is required, and personal computer clusters are not effective.
Human health is maintained by the properly balanced work of many symbiotic microbes in the body. In addition, the balance is considered to have several locally stable points. Comprehensive sequencing of genome fragments of microorganism gastro-intestinal which exist in the body's internal environment including oral, digestive, skin and urogenital system has been implemented for about 500 people in the Human Microbiome Project (HMP) in the USA and for about 300 people in the MetaHIT Project in Europe. However, when analyzing these valuable data, the rate of non-analyzable sequence fragments to be excluded will be significantly increased only with insensitive methods hypothesizing highly similar character strings.
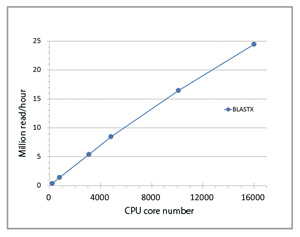
We firstly established the super parallel execution environment of the existing BLASTX software with the cooperation of Prof. Ken Kurokawa (Tokyo Institute of Technology), who is an expert in genomic analysis. A scalability up to 16,000 cores was confirmed on TSUBAME 2.0 of the Tokyo Institute of Technology (Fig. 1). Binary-tree transfer was used for applying the bandwidth of networks at the initial delivery of the database to each calculation node. After translating DNA fragment sequences into amino acid sequences in six possible frames, we flexibly compared them with existing amino acid sequences. Event though the target DNA sequence fragments mutated at the DNA sequence level, they are often consistent with residues of existing sequences at the amino acid sequence level, or are changed into residues with similar physicochemical properties. Therefore, our flexible method which compares using a distance matrix between characters improves sensitivity for sequence search to explore distant genes with the same functions.
We then created a software, GHOSTX, as a substitute for BLASTX, with drastic speedup on the algorithm [1, 2]. GHOSTX can find similar short strings between database and query sequences immediately by multito-multi comparison by describing a partitioned database stored in memory using Suffix Array, translating a query sequence to Suffix Array, and comparing theme ffectively (Fig. 2). While BLASTX compares only character strings with a fixed-length, charac ter strings for comparison canbe extended with variable-length until they satisfy a similarity threshold score in GHOSTX, whic h improves the sensitivity per calculation cost. GHOSTX achieved about 20 times processing speed even when an equivalent sensitivity an equivalent sensitivity with BLASTX was required. When the sensitivity is decreased to a degree of no difficulty, GHOSTX attained over 100 times the speed. Its sensitivity is much better and it is 2 to 3 times faster than the BLAT algorithm which had high speed but was not able to be used because of poor sensitivity. We established a super parallel analysis system named "GHOST-MP" by implementing the GHOSTX algorithm on the ten quadrillion speed computer "K computer ", and integrating thread parallelization using OpenMP and data parallelism between nodes using MPI.
For the principal parts of homologous search, good scalability up to 1,536 nodes (12,288 cores) was measured with "K computer". Although the execution time for each sequence fragment varies widely, a scalability of up to several tens of thousands of nodes is expected because of the load balancing mechanism. In GHOST-MP, I/O operation is not completely intensive because its calculations are proportional to the product (multiplication) of length of the database and length of query sequence sets. Even though, careful I/O design is required. In the case of "K computer", I/O transfer to and from I/O nodes takes place in the z axis direction. We aim to achieve highly- efficient implementation up to several tens of thousands of nodes by adjusting data input using representative nodes and a broadcast mechanism, and by improving dispersion and reduction of I/O load.
References
[1] Suzuki, Ishida, Akiyama (2010) IPSJ Technical report, 2010-BIO-23(21):1-6
[2] Suzuki, Ishida, Akiyama (2011) IPSJ Technical report, 2011-BIO-25(32):1-8
 |
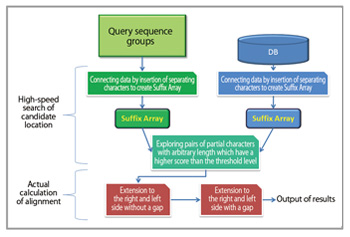 |
Fig 1 : Parallel performance in |
Fig 2 : Processing flowchart in GHOSTX algorithm |
BioSupercomputing Newsletter Vol.5
- SPECIAL INTERVIEW
- The time has come for biosupercomputing to get results with the world's No. 1 supercomputer
"K computer", and take up the challenge of "prognostic biology".
Deputy Program Director of Computational Science Research Program, RIKEN Ryutaro Himeno - What should we do to promote industrial use of sophisticated computer resources and development applications?
Chief Coordinator of Foundation for Computational Science Masahiro Fukuda
Chief Researcher of Urban Innovation Institute and Executive Board Member and Bureau Chief of BioGrid Center Kansai Ryuichi Shimizu
- Report on Research
- Analysis of molecular mechanism of enzymatic reactions by QM/MM Free Energy Method
Graduate School of Science, Kyoto University Shigehiko Hayashi (Molecular Scale WG) - Computational Mechanobiology of Actin Cytoskeleton
Institute for Frontier Medical Science, Kyoto University Yasuhiro Inoue (Cell Scale WG) - Development of Blood flow Analysis Method for Simulation of Thrombus Formation
Department of Mechanical Engineering, The University of Tokyo Satoshi Ii (Organ and Body Scale WG) - Development of Data Assimilation Technology for Simulation of Living Things
The Institute of Statistical Mathematics Tomoyuki Higuchi (Data Analysis Fusion WG)
- SPECIAL INTERVIEW
- Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Program Director of RIKEN HPCI Program for Computational Life Sciences Toshio Yanagida
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences Akinori Kidera
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences Yukihiro Eguchi
- Report on Research
- Simulation Applicable to Drug Design
Research Center for Advanced Science and Technology, The University of Tokyo Hideaki Fujitani (Field 1- Program 2) - An Ultra-fast Analysis System for Next-Generation DNA Sequencer Data
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
Yutaka Akiyama, Takashi Ishida, Masanori Kakuta, Shuji Suzuki (Field 1- Program 4)
- Report
- BioSupercomputing Summer School 2011
Computational Science Research Program, RIKEN Yasuhiro Ishimine (Organ and Body Scale WG)
Research and Development Center for Data Assimilation, Institute of Statistical Mathematics Masaya Saito (Data Analysis Fusion WG)
Niigata University of International and Information Studies Eisuke Chikayama (Cell Scale WG)
School of Medicine, Tokai University Yohei Nanazawa (Cell Scale/Organ and Body Scale WG)
Computational Science Research Program, RIKEN Takashi Handa (Brain and Neural Systems WG)
Computational Science Research Program, RIKEN Gen Masumoto (High-performance Computing Team)
Computational Science Research Program, RIKEN Kei Moritsugu (Molecular Scale WG) - “Next-Generation Integrated Simulation of Living Matter (ISLiM)”, a web page dedicated to new applications, has opened.
Computational Science Research Program Integrated Simulation of Living Matter Group
- Event information