

Simulation Applicable to Drug Design
HPCI Strategic Program Field 1 Supercomputational Life Science

Research Center for Advanced Science and Technology, The University of Tokyo
Hideaki Fujitani (Field 1- Program 2)
The examination of interaction between a protein and a compound thatspecifically binds to the protein (ligand) is not only a major challenge inthe basic study of life phenomena, but also extremely important from theaspect of application. Accurate evaluation of the free energy of bindingbetween proteins and drug molecules is a longstanding challenge in thefield of drug discovery. Although various methods including Docking-Simulation have been tried, there has been no method that predicts thefree energy of binding to a protein with an accuracy effective enoughfor designing a drug molecule. For finding the difference in free energybetween two different states of thermal equilibrium, one in which theprotein and its ligand bind together in water (cell fluid) and the other inwhich they separately dissolve in water, a methodfor accurate incorporation of the entropy changedue to conformational change of the proteinand molecular motion is required. For such acalculation, the molecular dynamics method isused and its accuracy depends strongly on the force-field parameter used.
Torsion interaction relative to the Ramachandranangle of the protein main chain is the most important force-field parameter in defining thecubic structure of proteins, and its energy profiles in a glycine peptide are compared in Fig 1[1]. The black solid line shows the profile defined through a molecular orbital calculation which is currently the most accurate (LCCSD). It is compared with conventional molecular force fields such as AMBER(ff99, ff03) and OPLS-AA. Ff99sb is a force field in which only the main chain torsion interaction of ff99 released in 2006 was modified, and defined so that it conforms to energy by high-accuracy molecular orbital calculations at some angles. For this reason, the energy values are close to those shown by the black line when φ and ψare 0 degrees. However when φ is 80 to 180 degrees, an energy barrier higher by 1kcal/mol is observed compared to the energy shown by the black line. By use of the electric charge of AMBER and the van der Waals parameter, we developed a method to assign force-field parameters in a unified manner to proteins, DNA, RNA or arbitrary compounds, and named it the FUJI force-field[2].
With a relational expression between the free energy difference ΔG and non-equilibrium work W[3], we performed molecular dynamics calculations independently at multiple intermediate states from the state in which there is full interaction between the ligand and the other molecule to the state in which there is no interaction between them[4], and calculated the free energy from the Work W required for shifting to the next stage (Fig2). With the MP-CAFEE method and the FUJI force field, it is possible to perform an accurate computation of proteinligand binding free energy.
[1] H. Fujitani, A. Matsuura, S. Sakai, H. Sato, and Y. Tanida : J. Chem. Theory Comput., 5, 1155 (2009).
[2] H. Fujitani, Y. Tanida, and Azuma Matsuura : Phys. Rev. E, 79, 021914 (2009).
[3] C. Jarzynski : Phys. Rev. Lett., 78, 2690-2693 (1997).
[4] H. Fujitani, Y. Tanida, M. Ito, G. Jayachandran, C. D. Snow, M. R. Shirts, E. J.Sorin, and V. S. Pande : J. Chem. Phys., 123, 084108 (2005).
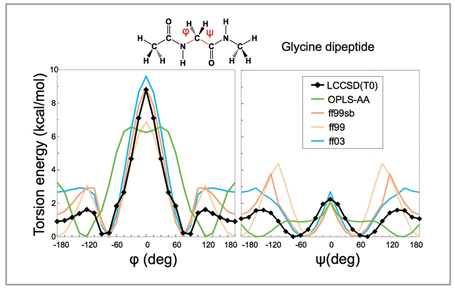 |
Fig 1 : Comparison of torsion interaction of protein main chain |
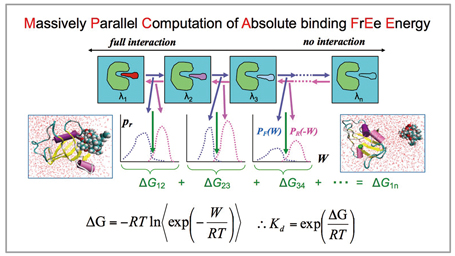 |
Fig 2 : Massively parallel computation of absolute binding free energy, MP-CAFEE |
BioSupercomputing Newsletter Vol.5
- SPECIAL INTERVIEW
- The time has come for biosupercomputing to get results with the world's No. 1 supercomputer
"K computer", and take up the challenge of "prognostic biology".
Deputy Program Director of Computational Science Research Program, RIKEN Ryutaro Himeno - What should we do to promote industrial use of sophisticated computer resources and development applications?
Chief Coordinator of Foundation for Computational Science Masahiro Fukuda
Chief Researcher of Urban Innovation Institute and Executive Board Member and Bureau Chief of BioGrid Center Kansai Ryuichi Shimizu
- Report on Research
- Analysis of molecular mechanism of enzymatic reactions by QM/MM Free Energy Method
Graduate School of Science, Kyoto University Shigehiko Hayashi (Molecular Scale WG) - Computational Mechanobiology of Actin Cytoskeleton
Institute for Frontier Medical Science, Kyoto University Yasuhiro Inoue (Cell Scale WG) - Development of Blood flow Analysis Method for Simulation of Thrombus Formation
Department of Mechanical Engineering, The University of Tokyo Satoshi Ii (Organ and Body Scale WG) - Development of Data Assimilation Technology for Simulation of Living Things
The Institute of Statistical Mathematics Tomoyuki Higuchi (Data Analysis Fusion WG)
- SPECIAL INTERVIEW
- Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Program Director of RIKEN HPCI Program for Computational Life Sciences Toshio Yanagida
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences Akinori Kidera
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences Yukihiro Eguchi
- Report on Research
- Simulation Applicable to Drug Design
Research Center for Advanced Science and Technology, The University of Tokyo Hideaki Fujitani (Field 1- Program 2) - An Ultra-fast Analysis System for Next-Generation DNA Sequencer Data
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
Yutaka Akiyama, Takashi Ishida, Masanori Kakuta, Shuji Suzuki (Field 1- Program 4)
- Report
- BioSupercomputing Summer School 2011
Computational Science Research Program, RIKEN Yasuhiro Ishimine (Organ and Body Scale WG)
Research and Development Center for Data Assimilation, Institute of Statistical Mathematics Masaya Saito (Data Analysis Fusion WG)
Niigata University of International and Information Studies Eisuke Chikayama (Cell Scale WG)
School of Medicine, Tokai University Yohei Nanazawa (Cell Scale/Organ and Body Scale WG)
Computational Science Research Program, RIKEN Takashi Handa (Brain and Neural Systems WG)
Computational Science Research Program, RIKEN Gen Masumoto (High-performance Computing Team)
Computational Science Research Program, RIKEN Kei Moritsugu (Molecular Scale WG) - “Next-Generation Integrated Simulation of Living Matter (ISLiM)”, a web page dedicated to new applications, has opened.
Computational Science Research Program Integrated Simulation of Living Matter Group
- Event information