

Analysis of molecular mechanism of enzymatic reactions
by QM/MM Free Energy Method

Graduate School of Science, Kyoto University
Shigehiko Hayashi (Molecular Scale WG)
Our group is developing the QM/MM free energy method for analyzing the molecular mechanism of enzyme reactions. Many molecular functions of biomolecules are coupled to chemical reactions with enzyme catalytic activity. Therefore, understanding the mechanism of enzymatic chemical reactions is important in control and design of biological molecular functions. The mainstream method for this purpose is currently the hybrid QM/MM approach in which quantum mechanics (QM) method is combined with the molecular mechanics (MM) method. In this method, very high computational efficiency is attained by describing the local active site of the enzyme catalyst by the QM method, and the other vast protein environment by the computationally cheap MM method. So far our group has elucidated molecular mechanisms of various enzyme reactions such as photoreceptor and molecular motor proteins by using the QM/MM approach.
In spite of the usefulness of the QM/MM approach, the calculation of enzymatic chemical reactions coupled to molecular function still remained difficult. In many cases, molecular function appears with accompanying by large conformational changes in biomolecules. Such conformational change is usually analyzed by molecular dynamics (MD) simulation based on the MM method. However, calculation of enzymatic chemical reactions coupled to conformational change requires a description based on the QM/MM method. In the QM/MM approach, however, QM calculation is computationally much more expensive compared to MM calculation, even though its application is limited to local active sites. Therefore, it is impossible to obtain enough MD sampling time to adequately take into account the slow relaxation of the biomolecular system.
In order to solve this problem, we have developed a novel QM/MM free energy method (QM/MM-RWFE-SCF method). In the QM/MM free energy method, the optimum free energy structure of the active site molecule treated by the QM method is determined on a free energy surface defined by the structural distribution of the MM region sampled by MD simulation. We developed a highly accurate and very efficient method by combining a mean field approximation with a statistical reweighting method, and then considering appropriately long-range coulomb interaction between QM and MM by the Ewald method. Especially in the scheme of this method, the calculations of the QM/MM method part are completely separate from the MD simulation part for MM structure sampling. Therefore, existing sophisticated MD programs can be applied to the MD simulation, which achieves great flexibility in performing calculations.
As a test of the method, it was applied to hydrolysis of the glycoside bond ofα-amylase shown in the reaction scheme in Fig 1. The protein system in water represented by a periodic boundary condition consists of 68,000 or more atoms, and QM molecules of the active site are described with abundant base functions of over 600 bases. By using this method, we determined the optimum energy structures of the reactant and the product of the reaction. In the result, as shown in Fig 2, their optimum structures were determined by the accompanying 90- and 21-ns large structural relaxation of protein loops surrounding active sites, respectively. Such a QM/MM free energy structural optimization calculation close to sub-microseconds has no precedent. More specifically, the structural optimization calculation performed by this method can follow the relaxation process of a protein structure ten-thousand times longer compared to direct QM/MM MD calculation, and describe relaxation 100 to 1000 times longer compared to other similar QM/MM free energy calculations. In addition, this calculation suggests the possibility that adjacent loops assume different structures (Fig 2). Such a major change of protein structure correlating with the enzyme reaction is a finding which cannot be obtained through conventional methods. It is believed to offer a new avenue for elucidating, controlling and designing new molecular mechanisms of enzyme reactions.
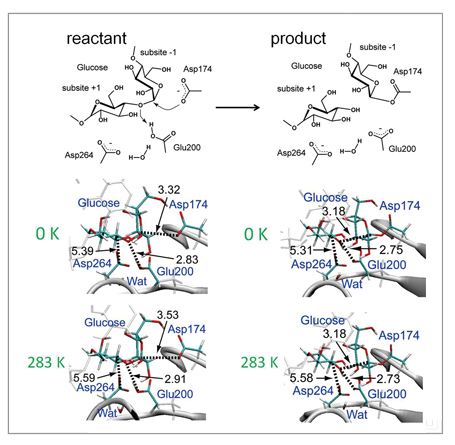 |
Fig 1 : Enzyme reaction scheme of α-amylase (above), and structural change from reactant to product (middle and below). Many parts of optimized structure on the free energy surface are different from the structure on the potential energy surface (0K), but it is also observed that the distance between atoms which have strong electronic interaction in the product is maintained in a thermal fluctuation. |
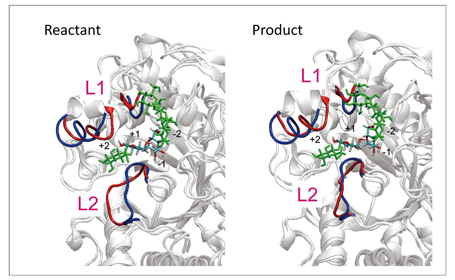 |
Fig 2 : Change of protein structure between reactant and product. The structure of the loops adjacent to active sites (L1 and L2 in red) obtained by free energy structure optimization are largely different from the structure before structural optimization (in blue). The structure of the L2 loop differs substantially between reactant and product. |
BioSupercomputing Newsletter Vol.5
- SPECIAL INTERVIEW
- The time has come for biosupercomputing to get results with the world's No. 1 supercomputer
"K computer", and take up the challenge of "prognostic biology".
Deputy Program Director of Computational Science Research Program, RIKEN Ryutaro Himeno - What should we do to promote industrial use of sophisticated computer resources and development applications?
Chief Coordinator of Foundation for Computational Science Masahiro Fukuda
Chief Researcher of Urban Innovation Institute and Executive Board Member and Bureau Chief of BioGrid Center Kansai Ryuichi Shimizu
- Report on Research
- Analysis of molecular mechanism of enzymatic reactions by QM/MM Free Energy Method
Graduate School of Science, Kyoto University Shigehiko Hayashi (Molecular Scale WG) - Computational Mechanobiology of Actin Cytoskeleton
Institute for Frontier Medical Science, Kyoto University Yasuhiro Inoue (Cell Scale WG) - Development of Blood flow Analysis Method for Simulation of Thrombus Formation
Department of Mechanical Engineering, The University of Tokyo Satoshi Ii (Organ and Body Scale WG) - Development of Data Assimilation Technology for Simulation of Living Things
The Institute of Statistical Mathematics Tomoyuki Higuchi (Data Analysis Fusion WG)
- SPECIAL INTERVIEW
- Pioneering the Future of Computational Life Science toward Understanding and Prediction of Complex Life Phenomena
Program Director of RIKEN HPCI Program for Computational Life Sciences Toshio Yanagida
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences Akinori Kidera
Deputy- Program Director of RIKEN HPCI Program for Computational Life Sciences Yukihiro Eguchi
- Report on Research
- Simulation Applicable to Drug Design
Research Center for Advanced Science and Technology, The University of Tokyo Hideaki Fujitani (Field 1- Program 2) - An Ultra-fast Analysis System for Next-Generation DNA Sequencer Data
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
Yutaka Akiyama, Takashi Ishida, Masanori Kakuta, Shuji Suzuki (Field 1- Program 4)
- Report
- BioSupercomputing Summer School 2011
Computational Science Research Program, RIKEN Yasuhiro Ishimine (Organ and Body Scale WG)
Research and Development Center for Data Assimilation, Institute of Statistical Mathematics Masaya Saito (Data Analysis Fusion WG)
Niigata University of International and Information Studies Eisuke Chikayama (Cell Scale WG)
School of Medicine, Tokai University Yohei Nanazawa (Cell Scale/Organ and Body Scale WG)
Computational Science Research Program, RIKEN Takashi Handa (Brain and Neural Systems WG)
Computational Science Research Program, RIKEN Gen Masumoto (High-performance Computing Team)
Computational Science Research Program, RIKEN Kei Moritsugu (Molecular Scale WG) - “Next-Generation Integrated Simulation of Living Matter (ISLiM)”, a web page dedicated to new applications, has opened.
Computational Science Research Program Integrated Simulation of Living Matter Group
- Event information