

QM/MM 自由エネルギー法による酵素反応分子機構の解析

京都大学大学院理学研究科
林 重彦(分子スケールWG)
我々のグループは、酵素反応分子機構の解析のためのQM/MM自由エネルギー法の開発を行っています。生体分子の分子機能は、多くの場合に酵素触媒活性を持つ化学反応と共役しています。従って、酵素化学反応機構の理解は、生体分子機能の制御及び設計に重要となります。その目的のために、現在、主流となっている手法が、量子化学的(QM)手法、及び分子力学的(MM)手法を組み合わせるハイブリッドQM/MM法です。この手法では、局所的な酵素触媒活性部位をQM法、及びその他の広大なタンパク質環境を計算コストの低いMM法で記述することにより、非常に高い計算効率を得ています。これまでに我々のグループはQM/MM法を用いて光受容や分子モータータンパク質等の様々な酵素反応の分子機構を解明してきました。
QM/MM法の有用性の一方で、分子機能に共役する酵素化学反応の計算は、依然困難でした。多くの場合、分子機能発現には生体分子の大きな構造変化が伴います。このような構造変化は、通常MM法に基づく分子動力学(MD)シミュレーションにより解析されますが、その構造変化に共役する酵素化学反応の計算にはQM/MM法に基づく記述が必要となります。しかしながら、QM/MM法では、局所的な活性部位のみの適用に制限したQM計算であってもMM計算に比べて圧倒的に計算コストが高く、生体分子系の遅い緩和を適切に考慮できるだけのMDサンプリング時間を得ることができません。
この問題を解決するために、我々は、新規なQM/MM自由エネルギー法(QM/MM-RWFE-SCF法)を開発しました。QM/MM自由エネルギー法では、MDシミュレーションによりサンプルされたMM部分の構造分布により定義される自由エネルギー曲面上で、QM法によって取り扱われる活性部位分子の最適自由エネルギー構造が決定されます。我々は、平均場近似と統計的reweightingの手法を組み合わせ、さらにQM-MM間の長距離クーロン相互作用をEwald法により適切に考慮することにより、非常に精度の高い効率的な手法を開発しました。特に、本手法のスキームでは、QM/MM法部分の計算とMM構造サンプリングのためのMDシミュレーション部分が完全に分離されます。従ってMDシミュレーションに既存の洗練されたMDプログラムを使用することが可能になり、計算遂行上の大きな柔軟性を達成しています。
開発された手法のテストとして、図1の反応スキームに示されるα-amylaseのグリコシド結合の加水分解に適用しました。周期境界条件で表される水中のタンパク質系は68,000以上の原子で構成され、活性部位のQM分子は600基底以上の豊富な基底関数を用いて記述されています。本手法を用いて、この反応の始状態及び終状態の自由エネルギー最適構造を決定しました。その結果、図2に示されるように、これらの最適構造は、活性部位周辺のタンパク質ループ部分のそれぞれ90ns及び21nsにわたる大きな構造緩和を伴い決定されました。このような、サブマイクロ秒に迫るQM/MM自由エネルギー構造最適化計算は前例のないものです。すなわち、本手法で行う構造最適化計算は、直接的なQM/MM MD計算に比べ、1万倍の長さのタンパク質構造緩和過程を追うことができ、また、他の同様なQM/MM自由エネルギー計算に比べても100〜1000倍程度の長時間緩和を記述しています。さらに、この計算により、反応前後において、近接ループが異なる構造を取る可能性が示唆されています(図2)。このような、酵素反応に相関するタンパク質構造の大きな変化は、これまでの手法では得ることのできない知見であり、酵素反応の新たな分子機構の解明や、制御・設計への道を拓くものと考えています。
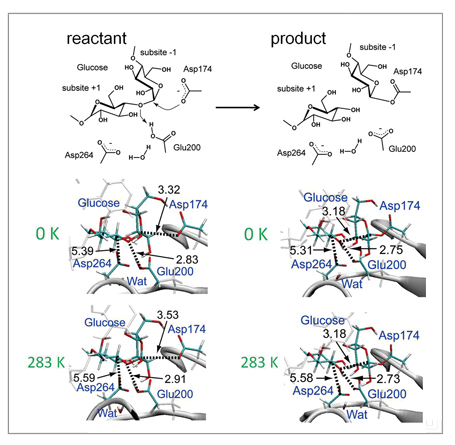 |
図1:α-amylaseの酵素反応スキーム(上図)と反応始状態(reactant)と終状態(product)の構造変化(中・下図)。自由エネルギー曲面上の構造最適化構造はポテンシャルエネルギー曲面上(0K)でのそれから変化している部分も多いが、終状態で強い電子的相互作用をしている原子間距離は、熱ゆらぎの効果を取り込んでも保持されていることがわかる。 |
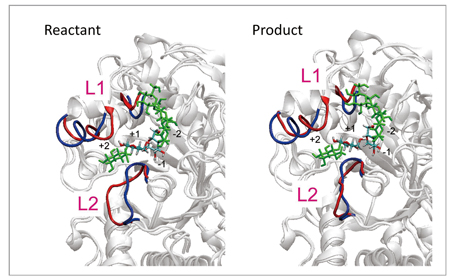 |
図2:反応始状態(reactant)と終状態(product)でのタンパク質構造の変化。自由エネルギー構造最適化により得られた活性部位近傍のループ(L1及びL2)の構造(赤)が、構造最適化前(青)の構造から大きく変化している。また、L2ループの構造は、始状態と終状態で大きく異なる。 |
BioSupercomputing Newsletter Vol.5
- SPECIAL INTERVIEW
- “予測する生物学”をめざすバイオスーパーコンピューティングの挑戦はいよいよ世界一の「京」で成果を出すフェーズに入った
理化学研究所 次世代計算科学研究開発プログラム 副プログラムディレクター 姫野 龍太郎 - 高性能計算機資源および開発アプリケーションの産業利用促進を図るために何をすべきか
計算科学振興財団 チーフコーディネーター 福田 正大
都市活力研究所 主席研究員 バイオグリッドセンター関西 理事・事務局長 志水 隆一
- 研究報告
- QM/MM 自由エネルギー法による酵素反応分子機構の解析
京都大学大学院理学研究科 林 重彦(分子スケールWG) - アクチン細胞骨格の計算メカノバイオロジー
京都大学再生医科学研究所 井上 康博(細胞スケールWG) - 血栓シミュレーションに向けた血流解析手法の開発
東京大学工学系研究科 伊井 仁志(臓器全身スケールWG) - 生命体シミュレーションのためのデータ同化技術の開発
統計数理研究所 樋口 知之(データ解析融合WG)
- SPECIAL INTERVIEW
- 複雑な生命現象の理解と予測に向けて計算生命科学の明日を拓く
理化学研究所 HPCI計算生命科学推進プログラム プログラムディレクター 柳田 敏雄
理化学研究所 HPCI計算生命科学推進プログラム 副プログラムディレクター 木寺 詔紀
理化学研究所 HPCI計算生命科学推進プログラム 副プログラムディレクター 江口 至洋
- 研究報告
- 創薬応用シミュレーション
東京大学先端科学技術研究センター 藤谷 秀章(分野1-課題2) - 次世代DNAシークエンサデータの超高速解析
東京工業大学大学院情報理工学研究科
秋山 泰 / 石田貴士 / 角田将典 / 鈴木脩司(分野1-課題4)
- 報告
- バイオスーパーコンピューティングサマースクール2011
理化学研究所 次世代計算科学研究開発プログラム
石峯 康浩(臓器全身スケールWG)
統計数理研究所 データ同化研究開発センター
斎藤 正也(データ解析融合WG)
新潟国際情報大学
近山 英輔(細胞スケールWG)
東海大学医学部内科学系循環器内科
七澤 洋平(細胞スケール/臓器全身スケールWG)
理化学研究所 次世代計算科学研究開発プログラム
半田 高史(脳神経系WG)
理化学研究所 次世代計算科学研究開発プログラム
舛本 現(開発・高度化T)
理化学研究所 次世代計算科学研究開発プログラム
森次 圭(分子スケールWG) - 「次世代生命体統合シミュレーションソフトウェアの開発(ISLiM)」開発アプリケーション紹介ページ、オープン
次世代計算科学研究開発プログラム 次世代生命体統合シミュレーション研究推進グループ