

Data Analysis Fusion WG
Exhaustive Protein-Protein Interaction Network
Prediction by Using
MEGADOCK
Graduate School of Information Science and Engineering,
Tokyo Institute of Technology
(From left, above)Yutaka Akiyama, Yuri Matsuzaki,
Nobuyuki Uchikoga and Masahito Ohue

We are working on prediction of the protein-protein interaction (PPI), one of the important problems of systems biology, by the method of bioinformatics and parallel processing (Figure 1). Usually, the expected role of the computational method using physical chemistry on PPI analysis was to examine the configuration and affinity of interactions concerning the known one-to-one protein-protein interaction in detail. We then developed “MEGADOCK,” a novel PPI prediction system based on large-scale parallel calculation, and made it possible to predict a candidate pair of PPI exhaustively from a large amount of protein groups. This is expected to contribute to the discovery of new PPI by collaboration with experiments in the future.
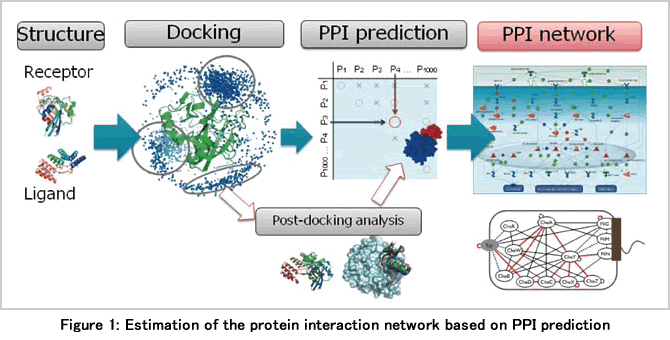
MEGADOCK is a system to predict the presence or absence of interaction using information about the tertiary structure of proteins based on various scores obtained from rigid-body docking. In this calculation, a high-speed evaluation is conducted mainly based on the shape complementarity of the molecular surface without considering the structural change of the protein. We introduced the rPSC (real Pairwise Shape Complementarity) score composed of the terms of shape complementarity and electrostatic interaction assigned to the molecular structures on the voxel space. With a conventional tool, ZDOCK, score is calculated using 3 interactions with 3 complex numbers, whereas in the rPSC, score is calculated using 2 interactions with 1 complex number by expressing the shape complementarity in a real number part and introducing electrostatic interactions into an imaginary number part. The number of three-dimensional fast Fourier transformation (FFT) required for convlolution sum calculation was reduced. When executed by a single CPU, about four times higher calculation speed was achieved with the same precision as ZDOCK.
MEGADOCK is parallelized using the MPI library. When a certain processor was assigned multiple receptor and ligand proteins, one ligand is taken sequentially from the ligand set, transformed by FFT with the certain angle increment, and compared as the innermost loop with all the data in the receptors set. A procedure of making the FFT transformed library concerning known proteins, and read from a hard disk to perform convolution sum calculations was implemented, and a speed increased of up to about 3 times was achieved. With appropriate load balancing, efficient calculation is possible by hundreds of processors or more.
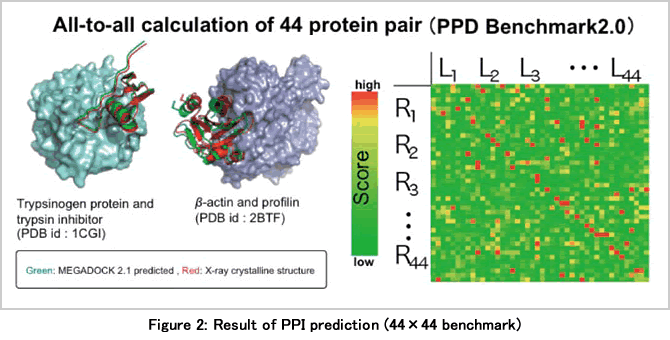
As a benchmarking of MEGADOCK, we firstly performed PPI prediction of 44×44=1,936 combinations on 44 protein complexes. The predicted (red) and native (green) structures were consistent (upper part of Figure 2). In the prediction of PPI pairs, many correct complexes were predicted as shown by the warm color on the diagonal line in the lower part of Figure 2, and a prediction performance (F-measure = 0.415) similar to or higher than in a related study was obtained. As an actual application in systems biology, PPI prediction was performed on the signal transduction pathway of bacterial chemotaxis (89×89=7,921) and the human EGFR signal transduction pathway related to lung cancer (497×497=247,009). Our goal is to perform the calculation of a 1,000×1,000 (mega) class routinely.
References
[1] Matsuzaki Y., Matsuzaki Y., Sato T. and Akiyama Y., J Bioinform Comput Biol , 7: 991-1012 (2009).
[2] Ohue, Matsuzaki, Matsuzaki, Sato and Akiyama. IPSJ Transactions on Mathematical Modeling and its Applications (TOM), 3(3): 91-106 (2010).
BioSupercomputing Newsletter Vol.3
- SPECIAL INTERVIEW
- The role of supercomputers is important for integrating various leading-edge research bases for proactive use.
Manager Clinical Development Planning and Management
Mochida Pharmaceutical Co., Ltd. Visiting Professor Tohoku University Kazumi Nishijima - Sonic simulation research in the body which is essential for promotion of ultrasound therapy and development
of therapeutic apparatus
Extraordinary researcher, Department of Mechanical Engineering, School of Engineering, University of Tokyo Akira Sasaki
- Report on Research
- Achievement of a Multiscale Molecule Simulation of QM/MD/CGM(Molecular Scale WG)
Institute for Protein Research, Osaka University
Yasushige Yonezawa / Shusuke Yamanaka / Hiromitsu Shimoyama / Hideki Yamazaki / Haruki Nakamura
RIKEN, Computational Science Research Program Ikuo Fukuda - Towards development and experimental demonstration of liver model based
on large-scale metabolic simulation at individual cellular level (Cell Scale WG)
School of Medicine, Keio University Ayako Yachie-Kinoshita - Exhaustive Protein-Protein Interaction Network Prediction by Using MEGADOCK (Data Analysis Fusion WG)
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
Yutaka Akiyama / Yuri Matsuzaki / Nobuyuki Uchikoga / Masahito Ohue - Whole Brain Simulation of the Insect Olfactory System
Research Center for Advanced Science and Technology, The University of Tokyo Tomoki Kazawa, Stephan Shuichi Haupt
- Report
- The summer school 2010 for the Integrated Simulation of Living Matter was held.
RIKEN, Computational Science Research Program Yasuhiro Ishimine (Organ and Body Scale WG)
The Institute of Medical Science, The University of Tokyo Teppei Shimamura (Data Analysis WG)
RIKEN, Computational Science Research Program Yasuhiro Sunaga (Cell Scale WG)
Kyoto University Graduate School of Informatics Naoki Honda (Brain and Neural WG)
RIKEN, Computational Science Research Program Gen Masumoto (High-Performance Computing Team)
RIKEN, Computational Science Research Program Kosuke Matsunaga (Molecular Scale WG) - After participation in the summer school 2010 for the Integrated Simulation of Living Matter
First year of doctor's course, The University of Tokyo Graduate School of Science Ken Saito