

Molecular Scale WG
Achievement of a Multiscale
Molecule Simulation of QM/MD/CGM
Institute for Protein Research, Osaka University
(From the above) Yasushige Yonezawa, Shusuke Yamanaka,
Hiromitsu Shimoyama, Hideki Yamazaki and Haruki Nakamura
RIKEN, Computational Science Research Program
Ikuo Fukuda

Our group is attempting to elucidate the mechanism of functions of biological macromolecules, proteins and nucleic acids, at multiple levels including molecular, atomic and electronic levels using molecular simulation. Actual molecules in living matter show thermally fluctuating actions at room temperature, and the fluctuation has the essential roles on molecular functions. Our purpose is to elucidate the mechanism of such biological molecules including their electronic state while simulating the thermal fluctuation in details.
For this achievement, we have developed a multiscale simulation program, Platypus (PLATform for dYnamic Protein Unified Simulation ), which couples quantum mechanics (QM) with molecular dynamics (MD) which were developed individually. Quantum mechanics to calculate electronic state is a computation method essential for handling enzymatic functions in simulation, and here, computation by the Hartree-Fock (HF) method, the density functional theory (DFT), CASSCF and CIS are available. Molecular dynamics is a simulation method, which reproduces the thermal fluctuation of molecules. Platypus is an integral of the program which realizes massive parallelism with the original codes, which has accelerated performance of up to 8192 parallels in the computations of both HF and DFT in the development so far, and particularly, it shows good scalability up to 1000 to 2000 parallels. In addition, Platypus loads the MD computation with an independently developed coarse graining model (CGM), with which the multiscale molecule simulation of QM/MD/CGM can be implemented. Moreover, we are developing an algorithm which performs efficient sampling with the QM/MD computation.
As one of the applications of Platypus , we introduce research on the cis-trans isomerization mechanism of the proline residue, which is related to the signal molecular control of various life activities. In a study of small peptides including proline in water (reference 1), the transition state between cis and trans forms the conventionally known pyramid as well as the inverse pyramid, and it was firstly shown that this state fluctuates between these 2 forms (Figure 1). From a study of the Pin1 isomerase, which accelerates the cis-trans isomerization of the proline residue, we succeeded in grasping the mechanism, in which the strain occurring by binding of the peptide including proline to the enzyme active site stabilizes the energy in the transition state so as to accelerate isomerization.
In the molecular dynamics simulation, the time for computation of interatomic long-range forces not based on chemical bonds between molecules accounts for a large amount. By parallelizing this part, the computation time can be considerably shortened. The Ewald method is a general method most often used, but it is known that this Ewald method is not suitable for parallelization of a very large system. We are therefore pursuing research to revise the long-range force potential recently developed by Wolf et al., and develop our original Force Switching-Wolf method (FSw-Wolf method), by which a consistent and steady simulation can be performed instead of the Ewald method. We succeeded in showing by simulation of a fused sodium salt (reference 2 and Figure 2) and a short peptide in water that the FSw-Wolf method can realize the computation at a precision comparable to that of the Ewald method. The FSw-Wolf method is not only a very convenient algorithm, but also it considers only the force between atoms at a relatively short distance. Thus, it is very suitable for large scale parallelized molecular dynamics simulation. Moreover, it is considered that this method will become a key method which can realize highspeed parallel computation by combining with other algorithms.
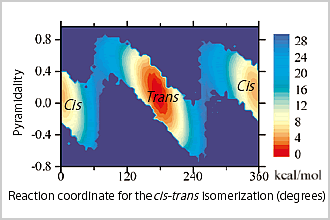
Figure1: Free energy landscape of cis-trans isomerization of a peptide including a proline residue, which was elucidated by QM/MD multiscale simulation. A transition state largely fluctuating between cis and states is observed. |
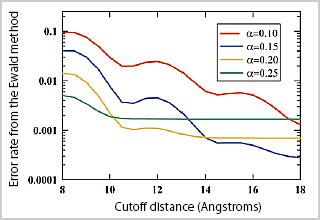
Figure2: The energy error between the FSw-Wolf method and the Ewald method, plotted as a function of the cutoff distance and the parameter α in the FSw-Wolf method. This shows that the FSw-Wolf method can reproduce an energy very close to that in the Ewald method. |
Reference 1. Yonezawa Y., Nakata K., Sakakura K., Takada T., Nakamura H., J. Am. Chem. Soc. 131(12), 4535-4540.
Reference 2. Fukuda I., Yonezawa Y., Nakamura H., J. Phys. Soc. Jpn. 77(11), 114301, 2008.
BioSupercomputing Newsletter Vol.3
- SPECIAL INTERVIEW
- The role of supercomputers is important for integrating various leading-edge research bases for proactive use.
Manager Clinical Development Planning and Management
Mochida Pharmaceutical Co., Ltd. Visiting Professor Tohoku University Kazumi Nishijima - Sonic simulation research in the body which is essential for promotion of ultrasound therapy and development
of therapeutic apparatus
Extraordinary researcher, Department of Mechanical Engineering, School of Engineering, University of Tokyo Akira Sasaki
- Report on Research
- Achievement of a Multiscale Molecule Simulation of QM/MD/CGM(Molecular Scale WG)
Institute for Protein Research, Osaka University
Yasushige Yonezawa / Shusuke Yamanaka / Hiromitsu Shimoyama / Hideki Yamazaki / Haruki Nakamura
RIKEN, Computational Science Research Program Ikuo Fukuda - Towards development and experimental demonstration of liver model based
on large-scale metabolic simulation at individual cellular level (Cell Scale WG)
School of Medicine, Keio University Ayako Yachie-Kinoshita - Exhaustive Protein-Protein Interaction Network Prediction by Using MEGADOCK (Data Analysis Fusion WG)
Graduate School of Information Science and Engineering, Tokyo Institute of Technology
Yutaka Akiyama / Yuri Matsuzaki / Nobuyuki Uchikoga / Masahito Ohue - Whole Brain Simulation of the Insect Olfactory System
Research Center for Advanced Science and Technology, The University of Tokyo Tomoki Kazawa, Stephan Shuichi Haupt
- Report
- The summer school 2010 for the Integrated Simulation of Living Matter was held.
RIKEN, Computational Science Research Program Yasuhiro Ishimine (Organ and Body Scale WG)
The Institute of Medical Science, The University of Tokyo Teppei Shimamura (Data Analysis WG)
RIKEN, Computational Science Research Program Yasuhiro Sunaga (Cell Scale WG)
Kyoto University Graduate School of Informatics Naoki Honda (Brain and Neural WG)
RIKEN, Computational Science Research Program Gen Masumoto (High-Performance Computing Team)
RIKEN, Computational Science Research Program Kosuke Matsunaga (Molecular Scale WG) - After participation in the summer school 2010 for the Integrated Simulation of Living Matter
First year of doctor's course, The University of Tokyo Graduate School of Science Ken Saito