

Development of Next-generation
Molecular Dynamics Simulation
Programs
High-performance Computing Team
(From left)Hiroshi KOYAMA, Yosuke OHNO, Gen MASUMOTO, Aki HASEGAWA, Gentaro MORIMOTO

The High-performance Computing team has been currently developing molecular dynamics simulation programs as basic applications for the next-generation supercomputer. Molecular dynamics is a calculation technique used to explain the motion of molecules, such as proteins, by solving Newton’s laws of motion in classical mechanics. Of course, the behavior of molecules would follow quantum mechanics in principle. The calculation of manybody quantum mechanics, however, requires a large number of calculations, even using the next-generation supercomputer. In addition, it is easier for us to understand the dynamic characteristics if we replace the motion with classical mechanics. This is why molecular dynamics is still an effective method. Molecular dynamics simulation can be one of the basic, classical research methods in the life sciences.
Then, why do we need to develop a new program? One reason is a serious requirement for scalability. The following might describe what scalability is in a simple way; "computation time is reduced by half when we use two computers." This may sound obvious, but it is essential for large-scale computing not to slow down performance, even if the number of computers is increased to a hundred or a thousand and so on. This means that we cannot create a supercomputer by just connecting cheap PCs. More importantly, scalability depends on not only the level of perfection of the hardware and software, but ultimately also the computational algorithm, since the purpose of scientific computing is to find a solution.
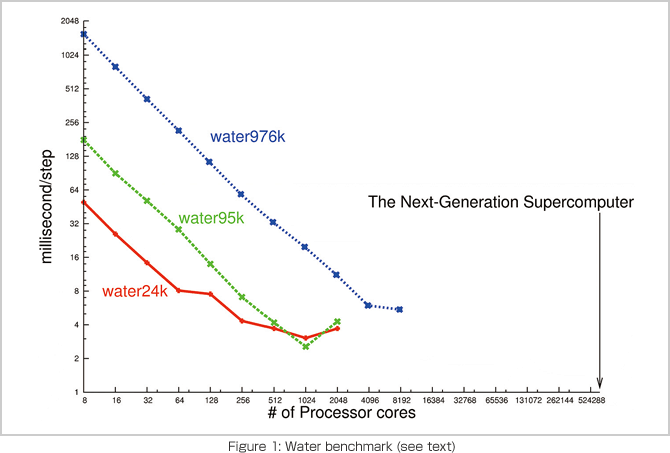
Figure 1 shows the run time of simulations using our molecular dynamics program for three different scales. This figure shows that computation time decreases in inverse proportion to the number of parallel computers, that is, it is scalable. On the other hand, every line has a plateau, while the number of parallel computers increases. This occurs when the communication time exceeds the computation time. In the graph, these plateaus occur at approximately 100 atoms per CPU core, regardless of the scale. This limit indicates the balance between the computing performance of a CPU and the performance of the data communication network. The number of parallel computers for the next-generation supercomputer is 100 times more than these simulations, as indicated by the arrow in Figure 1. To compute 100 times faster, we need to increase the communication speed 100 times, reduce the data amount to 1/100 or make the scale of matter 100 times larger. Since the communication speed depends on the hardware specifications, the improvement of software has a limitation. As the reduction of amount of data is a mathematical issue through an algorithm rather than a programming issue, we need to seek a new method. While this is hard to achieve, it is also interesting and challenging.
What can we do if large-scale molecular dynamics simulations become possible through the next-generation supercomputer? One application is drug discovery. Drugs are substances (keys) that bond closely to the surface of target proteins (keyholes) and develop chemical activities. Searching for and choosing these candidates is called "drug-discovery screening." The compound data used by conventional screening contains data on approximately eight million compounds in general. It has been pointed out that discovering new probable structures is already difficult. According to the theoretical estimation that assumes that approximately 1063 compounds satisfy drug likeness, however, screening has only been performed on a small percentage of these compounds. If we can make a virtual compound library consisting of one billion compounds that are likely to result in new structures, the possibility of discovering new drugs dramatically increases. The nextgeneration supercomputer and next-generation molecular dynamics programs will make this possible by increasing the speed of this task. The new library needs to calculate the assay indices for diversity, newness, drug likeness and assay availability by comparing the calculated indices to the indices in the conventional library. To achieve this, we are currently creating a prototype on the scale of 10 million to 100 million compounds (Figure 2).

Finally, we would like to thank the Advanced Center for Computing and Communication at RIKEN for letting us use the RIKEN Integrated Cluster of Clusters (RICC) system to execute the numerical simulations in this report.
BioSupercomputing Newsletter Vol.2
- SPECIAL INTERVIEW
- Aiming to Become a Global Trendsetter in the Life Sciences by Making the Best Use of the Next-generation Supercomputer!
Computational Science Research Program Deputy Program Director
Ryutaro HIMENO
- A Message from the Team Leader
- Simulating Cell Phenomena by Recreating Cells as They Exist in Living Organisms
Cell Scale Team Team Leader
Hideo YOKOTA - Create a Brain on a Supercomputer to Unravel the Functions of the Brain and Nervous System
Brain and Neural Systems Team Team Leader
Shin ISHII - High-performance Computing Environment to Maximize the Potential of the Next-generation Supercomputer
High-performance Computing Team Team Leader
Makoto TAIJI
- Report on Research
- Protein Reaction Simulation Based on All-electron Calculation
Institute of Industrial Science, the University of Tokyo
Fumitoshi SATO / Toshiyuki HIRANO / Noriko UEMURA / Naoki TSUNEKAWA / Junichi MATSUDA - Full Eulerian Fluid-structure Coupled Method
Associate Professor at School of Engineering, the University of Tokyo
Kazuyasu SUGIYAMA - A Large-scale Simulation Model of Cortical Microcircuits: CMDN(Cortical Microcircuit Developed on NEST)
Brain and Neural System Team
Jun IGARASHI - Development of Next-generation Molecular Dynamics Simulation Programs
High-performance Computing Team
Hiroshi KOYAMA / Yosuke OHNO / Gen MASUMOTO / Aki HASEGAWA / Gentaro MORIMOTO