(Updated on 2016/1/28)
理化学研究所ゲノム医科学研究センター 角田 達彦
ヒトのゲノム全体にわたる遺伝的な相違点を、患者群とコントロール群で比較することにより、疾患の遺伝的原因を探るための統計検定計算。
Haplotype頻度を用いたType I errorの確率計算
マルコフ連鎖モンテカルロ法
ハイブリッド並列
C, MPI
ソース・コードをISLiMダウンロードサイトから公開済み。
- 90人のデータを対象に、遺伝的変異の地域差の解析
- PCクラスタで8192コア並列
- メモリ容量 10 GB、ディスク容量 10 GB
- 約20万人分のデータを対象に、 約50疾患の解析
- 64万コア並列
- メモリ容量 2TB 、ディスク容量 2TB
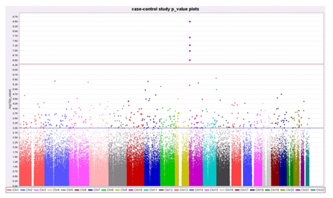
図. SNP遺伝子型と疾患との関連を示すアーミテージ検定のP値の -log P値
- 全ゲノム(約30億塩基)の個人毎の遺伝情報の違いの中から、疾病に関連する遺伝情報を網羅的に精度よく探し出すことが可能になる。
- ParaHaploソフトウェア講習会 -> リンク
理化学研究所ゲノム医科学研究センター 角田 達彦
次世代シークエンサーの出力データを高速に解析し、ヒト個人間の遺伝的差異やがんゲノムの突然変異を高い正確さで同定する。
ヒト標準ゲノム配列に対するマッピングと確率計算に基づいた多様性検出
直接法による密行列の対角化
領域分割
Perl, C
ソース・コードをISLiMダウンロードサイトから公開済み。
- 初の日本人ゲノムシークエンス解析、がんゲノム解析による突然変異同定をPCクラスタ2000コアで実行
- メモリ容量 4 TB、ディスク容量 100 TB
- 500人のがんゲノム配列解析による突然変異同定を64万コアで実行
- メモリ容量 2PB、ディスク容量 50 PB
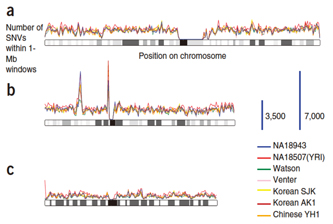
7人の全ゲノムシークエンスの比較
1Mbp当たりのSNP数 (a)1番染色体、(b)6番染色体、(c)X染色体 Nature Genetics 42, 931–936.
- 全ゲノム(約30億塩基)の個人毎の遺伝情報の違いを網羅的に精度よく検出
- がんの全突然変異を高速に検出し、創薬のターゲット分子を探索する。
- NGS Analyzerソフトウェア講習会 -> リンク
理化学研究所ゲノム医科学研究センター 角田 達彦
遺伝子間相互作用が発症リスクを変化させる疾患関連遺伝子の組合せを全ゲノムで探索する。2SNP間の全組合せを超並列に行う方法と、SNP間の連鎖不平衡(相関)も考慮した、より精密な方法の2種類を実装。前者で全組合せをスクリーニングし、後者で経験的p値を求める手順を想定している。
RAT(Rapid Association Test)
インポータンスサンプリング
データ分割
C++, MPI, OpenMP
ソースコードをISLiMダウンロードサイトから公開済み
- 10万SNP、4000人分のタイピングデータ、2SNP全組合せ
- 50億組合せ×4000人分のSNPデータ
- RICC 8192コア、メモリ容量 1.1 GB、ディスク容量 2 GB
- 全染色体上SNPの1万人規模、2SNP全組合せで解析
- 2500億組合せ×1万人×47疾患のSNPデータ
- メモリ容量 20 GB、ディスク容量 2 TB
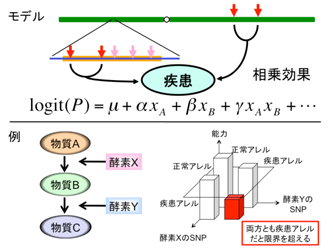
相互作用を起こして発症リスクを高める疾患関連遺伝子の探索
- 遺伝子どうしが相互作用を起こして疾患への発症リスクを上昇する現象とそれらの遺伝子を新たに疾患関連遺伝子として発見することができる。
東京大学医科学研究所教授 宮野悟
細胞内分子の発現制御システム(遺伝子ネットワーク)のモデル化・予測を行う超並列計算機用ソフトウェア群。
ノンパラメトリック回帰ベイジアンネットワーク、状態空間モデル、グラフィカルガウシアンモデル、ベクトル自己回帰モデル
発見的構造推定アルゴリズム+ブートストラップ法、Neighbor Node Sampling & Repeatアルゴリズム、並列最適構造推定アルゴリズム、EM法、L1正則化法
MPI,OpenMP, EP
Fortran90, C, R
ISLiMダウンロードサイトから公開済み
- 2万遺伝子からなる遺伝子ネットワーク
- RICC、ヒトゲノム解析センターの8192コアを使用
- メモリ容量 12 TB、ディスク容量 500 MB/network
- 全転写産物(10万以上)の遺伝子ネットワーク推定
- 64万コアを使用
- メモリ容量 1PB 、ディスク容量 10 GB/network
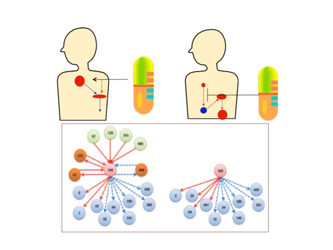
創薬ターゲット遺伝子のイン・シリコ探索
- ヒト全遺伝子産物を対象にした遺伝子ネットワーク推定による創薬ターゲット遺伝子のイン・シリコ探索が大規模にできる。
- ネットワークを用いて、被影響遺伝子の同定、作用点の推定、副作用の予測・回避、創薬ターゲット・毒性関与パスウェイ探索などが大規模に行える。
- 様々なデータから得られる様々な遺伝子ネットワークを超短時間で計算できるようになる。
- SiGN-BN講習会ビデオと配布資料 -> リンク
東京大学医科学研究所教授 宮野悟
データ解析融合プラットフォームで開発した技術とプログラムのうち、特にSiGN、LiSDASを解析パイプラインのコンポーネントとしてユーザが簡単に利用できる高機能GUIを備えたソフトウェアプラットフォームをユーザに提供。京に用意されているジョブスケジューリングシステムと連携し、京上で各解析パイプラインの処理の一部を実行し、その結果については、SBiPの視覚化コンポーネント群を用いて可視化し保存することが可能。
解析パイプラインのコンポーネントに依存
解析パイプラインのコンポーネントに依存
解析パイプラインのコンポーネントに依存
JAVA, R
ISLiMダウンロードサイトから公開済み
理研RICC上で作成したパイプラインをbatchジョブとして実行処理可能
解析パイプラインのコンポーネントに依存
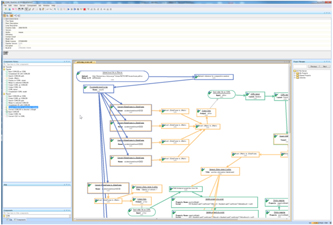
ネットワーク推定のための解析パイプラインの設計
- 用意されているさまざまな解析コンポーネントを自由に組み合わせて各解析者がカスタマイズした解析フローを実行可能。例えばSiGN-SSM、 SiGN-L1、SiGN-BNなどの遺伝子の発現情報からネットワーク推定を京上で推定するコンポーネント群を実行し、それらの結果をユーザに整理し視覚化して表示する解析プラットフォームを統合して利用できるようになる予定。
統計数理研究所モデリング研究系教授 樋口知之
生体内分子の計測データを参照値としてあたえることで、シミュレーションの再現性・予測力を改善するためのパラメータチューニングやモデルの改良を自動で行う。
差分法
階層化粒子フィルタ
ハイブリッド並列化。スレッド並列化が下層部をMPI並列化が上層部を担当する。
Fortran90, C, C++, MPI, OpenMP
ソース・コードをISLiMダウンロードサイトから公開済み
- 哺乳類の遺伝子概日周期発現に関わる転写制御モデルにおける力学パラメータ探索
- 100億粒子、未知パラメータ数44
- RICC8192コア、メモリ容量 3.5 TB、ディスク容量 16 TB
- 大規模パスウェイ・モデルにおけるパラメータ推定
- 500億粒子、未知パラメータ数250
- 320,000コア、メモリ容量 100 TB、ディスク容量 1 PB
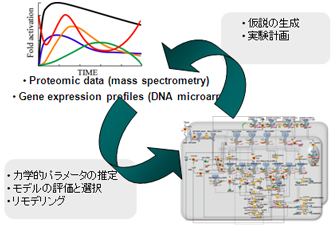
計算モデルのファインチューニング
- データ同化を行うことにより、未知パラメータを100~200個程度含む大規模パスウェイ・モデルのパラメータ推定が可能になる。
- さらに、自動生成した仮説モデルにデータ同化を行うことで、より再現・予測能力の高いモデルに再構築することが可能になる。
東京工業大学大学院情報理工学研究科教授 秋山泰
タンパク質間相互作用予測について、タンパク質立体構造情報から、表面形状相補性と静電相互作用に基づく単純化した評価モデルを新規に提案した。FFTを用いて計算量を大きく減じ、大規模並列計算機上で効率的な並列計算が可能。
二つの構造間の結合性を三次元複素畳込みで評価。
形状相補性と静電相互作用スコアを一つの複素数で表し、二体間の畳込み和をフーリエ空間上で行い逆FFTで評価値を得る。
- ノード内の回転角度並列: OpenMP
- ノード間の回転角度並列、データ並列: MPI
C++, MPI, OpenMP, FFTW3.2.2
ソース・コードをISLiMダウンロードサイトから公開済み
- ヒトEGFR系タンパク質の網羅的相互作用予測
(タンパク質500×500=250,000ペアの計算) - PCクラスタで512コア並列
- メモリ容量 1.5 TB (3GB/コア)、ディスク容量 250 TB (1GB/ペア)、実行時間 20,000,000秒
- ヒト肺がん関連シグナル伝達系タンパク質の網羅的相互作用予測 (タンパク質1,000×1,000級ペアの計算を様々な条件について行う。構造変化を考慮し10個×10個/ペア程度のアンサンブルを計算。)
- 40,000ノード、32万コア並列(8コア/ノード)
- メモリ容量 640 TB (2G/コア未満へ削減を仮定) 、ディスク容量 1 PB (主要部のみ保存)、実行時間1条件あたり100,000秒
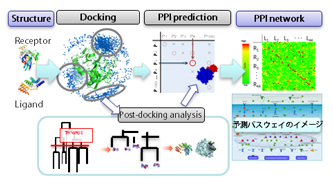
MEGADOCKによるPPIネットワーク予測の流れ
対象とするタンパク質の構造を入力すると、全組み合わせについて網羅的にドッキングを行い、結果のスコア分布を解析して、相互作用可能なタンパク質ペアを出力することでタンパク質間相互作用ネットワークを予測する。
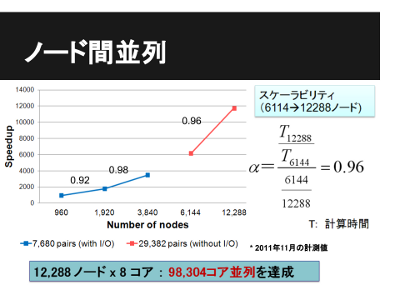
MEGADOCKのスケーラビリティ性能
- 例えばヒト細胞内では数万種類存在するといわれるタンパク質が相互にどのような制御関係にあるかを理解することができる。病因の解明や薬剤設計における重要な指針を与える。
- 「MEGADOCK-K」 ソフト講習会 -> リンク