(Updated on 2013/3/26)
Tatsuhiko TSUNODA, Center for Genomic Medicine, RIKEN
The ParaHaplo code is an invaluable parallel computing tool for conducting haplotype-based genome-wide association studies (GWAS) as the data sizes of projects continue to increase.
Exact probability calculation of type I error using haplotype frequencies.
Markov-chain Monte Carlo (MCMC) algorithm
Hybrid parallelization (Threads and MPI)
C and MPI
Source code is available through ISLIM download site.
- Analysis of regional differences in genetic variation for 90 people's data
- An 8,192 core parallel computing with RIKEN RICC cluster
- Required memory size/disk storage size: 10 GB/10 GB
- Analysis of almost 50 diseases for about 200,000-people's data
- parallel computing with 640-thousands of cores
- Required memory size/storage size: 2TB/2TB
Ref.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2774321/
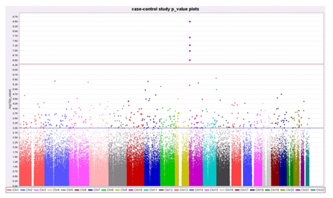
Figure 1. The figure shows -log (p-value) where the p-value in the Armitage test represents disease/SNP associations.
- Precise and comprehensive findings for disease associated genomic information among a variety of personal whole genomes (about 3 giga bases per person).
Tatsuhiko TSUNODA, Center for Genomic Medicine, RIKEN
The output data generated by a next-generation genome sequencer are analyzed in high speed by the NGS analyzer. The code identifies genetic differences among persons or cancer cell's mutations much precisely
Mapping for the human genome sequences and detection of diversities based on the probability calculations
Diagonalization of a dense matrix with a direct method
domain decomposition
Perl, C
Source code is available through ISLIM download site.
- The first Japanese genome sequence analysis and an identification of mutations in a cancer genome analysis using a 2,000-core x86 cluster system
- Required memory/disk: 4 TB/100 TB
- An identification of mutations in a 500-people's cancer genome analysis using 640 thousands of cores
- Required memory/disk storage: 2PB/50 PB.
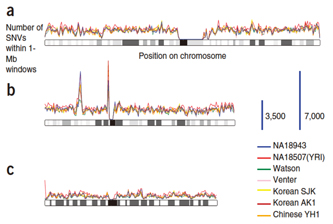
Figure 1. The comparisons among seven persons' whole genomes. The number of SNPs/Mbp is shown for (a) chromosome 1, (b)chromosome 6, and(c)chromosome X (Nature Genetics 42, 931–936)
- Comprehensive and precise detection of the differences among the personal genetic data (whole genome has about 3 billion bases per person)
- Find all of cancer's mutations faster and search target molecules for a drug discovery.
Tatsuhiko TSUNODA, Center for Genomic Medicine, RIKEN
- ExRAT exhaustively searches the genome for combinations of disease related genes/SNPs to identify significant disease association.
- The algorithm implements two methods - a massively parallel method for looking at all SNP-pairs, and a more precise method that takes the linkage disequilibrium between SNPs into account. The former picks up candidates, whereas the latter calculates empirical p-values.
RAT(Rapid Association Test)
Importance sampling
Data decomposition
C++, MPI, OpenMP
Source code is available through ISLIM download site.
- Genotyped data of 100,000 SNPs x 4,000 individuals
- Five billions of SNP-SNP combinations × 4,000 individuals
- RICC system with 8,192 cores
- Required memory/disk storage: 1.1 GB/2 GB
- Analysis on tens of thousands individuals (per one disease)
x 250 billion combinations of two SNPs in the human genome
x 50 diseases - Required memory/disk storage: 20 GB/2 TB
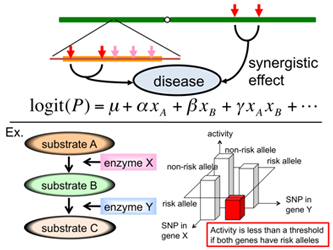
Figure 1. Searching the disease related genes that increase disease risks through epistatic interactions.
- ExRAT can identify gene-gene interactions that increase disease risk and identify novel disease related genes.
Satoru MIYANO, Professor, Institute of Medical Science, University of Tokyo
Massively parallel software series for modeling and estimation of a gene expression control system (genome network) in a cell
Nonparametric regression Bayesian networks, state space models, graphical Gaussian models, vector autoregressive models
Heuristic structure estimation algorithms + the bootstrap method, the neighbor node sampling & repeat algorithm, a parallel optimal structure estimation algorithm, the EM method、the L1 regularization method
MPI,OpenMP, EP
Fortran90, C, R
Code is available through ISLIM download site.
- A gene network having 20 thousands of genes
- RICC's 8,192-core and the supercomputer at the Human Genome Center
- Required memory/disk storage: 12 TB/500 MB (Need network)
- A gene network estimation for all transcripts (>100 thousands)
- Use 640 thousands of cores
- Required memory/disk storage: 1PB/10 GB (Need network)
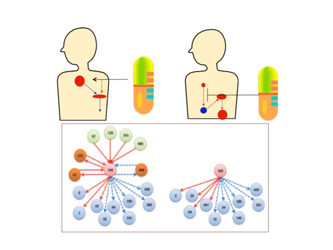
Figure 1.In silico search of the targeting genes for the drug discovery.
- A large scale in silico search of the targeting genes for the drug discovery using the method of the gene network estimation that will cover all human gene's transcripts
- Using the gene network, the identification of influenced genes, the estimation of active sites, the prediction and avoiding of side effects, the search of drug discovery targets and pathways being associated with toxicity can be done on a large scale
- A variety of gene networks can be solved in the shortest time. Reference: http://sign.hgc.jp/index.html
Satoru MIYANO, Professor, Institute of Medical Science, University of Tokyo
- The code provides a data analysis fusion platform that a user can use, in particular, SiGN and LiSDAS more easily among the developed codes for the data analysis fusion platform, combined with a highly functional and easy to use GUI.
- The K computer's job scheduling system supports to run a part of the analysis pipeline on the K computer, and a user can visualize and store the results using the SBiP's visualization component series
Depending on the components in analysis pipelines
Depending on the components in analysis pipelines
Depending on the components in analysis pipelines
JAVA, R
Code is available through ISLIM download site.
The pipeline created in RICC can be run as a batch job
Depending on the components in an analysis pipeline.
Figure 1. The design of the pipeline for a gene network estimation.
- By combining a variety of analysis components already being available, users can run their customized analysis flows.
- The analysis flow, for example, from a gene network estimation derived by gene expression information with SiGN-SSM, SiGN-L1, or SiGN-BN running on the K computer to a visualization after data reduction will be available.
Tomoyuki HIGUCHI, Professor, Institute of Statistical Mathematics
The software features basic functions to explore kinetic parameters in a biological pathway model as well as to reconstruct the graphical structure of an initial input simulator so that the reproducibility and predictivity of refined simulation models are improved for given experimental data on the endogenous variables.
Finite difference method
Hierarchical particle filter with two-layers
MPI for the upper layer, and OpenMP for the lower layer
Fortran90, C, C++, MPI, OpenMP
Source code is available through ISLIM download site.
- Estimation of kinetic parameters in a transcription control model for the mammalian circadian rhythm
- Particles: 10 billion, unknown parameters: 44
- RICC's 8,192 cores
- Required memory/disk storage: 3.5 TB/16 TB
- Parameter estimations in a large scale pathway model
- Particles: 50 billion, unknown parameters: 250
- Use 320,000 cores
- Required memory/disk storage: 100 TB/1 PB.
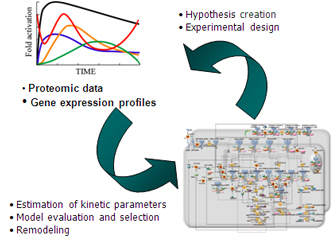
Figure 1. The fine tuning of a computational model.
- LiSDAS features basic functions to explore kinetic parameter values in a biological pathway model and to retrieve new hypothetical models (reconstruction of unreliable models) from a quite huge space of potential model sets such that simulation trajectories reproduce experimentally-observed profiles of model variables on diverse scales.
Yutaka AKIYAMA, Professor, Tokyo Institute of Technology
The code implements the simplified novel evaluation model for protein-protein interactions (PPI) based on the surface shape complementarily and the electrostatic interactions derived from the protein's 3D conformation data. Adopting FFT much reduces computing time to run faster on a large scale parallel computer
The complex convolutions on 3-D voxel models evaluate the binding between two protein conformations
The surface shape complementarities and the electrostatic interactions are scored with a single complex number. The convolution calculation for two conformations is done in Fourier space, and the inverse Fourier transformation gives the evaluated values.
OpenMP for intra-node and MPI for inter-node are applied to the rotation angle parallelization.
C++, MPI, OpenMP, FFTW3.2.2
Source code is available through ISLIM download site.
- Interaction prediction of human EGFR proteins (250,000 pairs)
- A 512-core x86 cluster (20,000,000 seconds)
- Required memory/disk storage: 1.5 TB/250 TB
- Interaction prediction for signal transduction pathway proteins relating human lung cancer (1000x1000 pairs for variable conditions. About 10x10/pair ensemble calculations corresponding to conformation change)
- Use 320 thousands of cores (100,000 sec/case)
- Required memory/disk storage: 640 TB/1 PB.
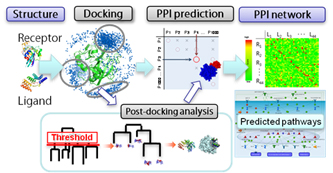
Figure 1. The flow of MEGADOCK's PPI network prediction. First, enter the target protein conformations. Second, the docking calculations are done for all combinations of the proteins. Third, the output score distributions are analyzed to get the protein pairs contributing to PPI. Fourth, the PPI network is predicted from the protein pair data.
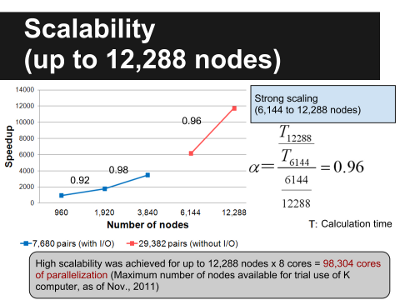
Figure 2. MEGADOCK Scalability
- The code can give, for example, understanding how several millions of proteins existing in a human cell mutually interact and control themselves. Such knowledge can give the important direction for drug discovery and resolving a disease cause.